 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePrediction of Hereditary Cancers Using Neural Networks
Jun 25, 2021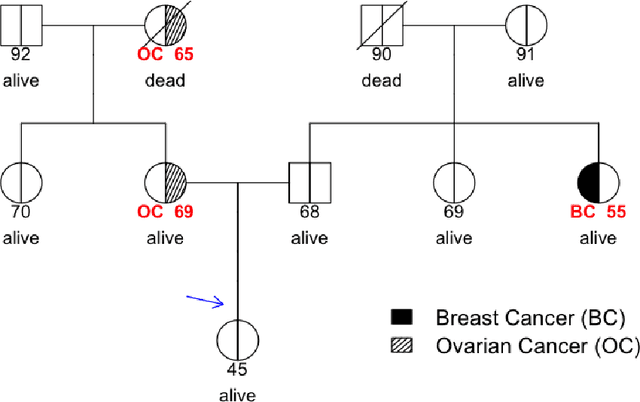
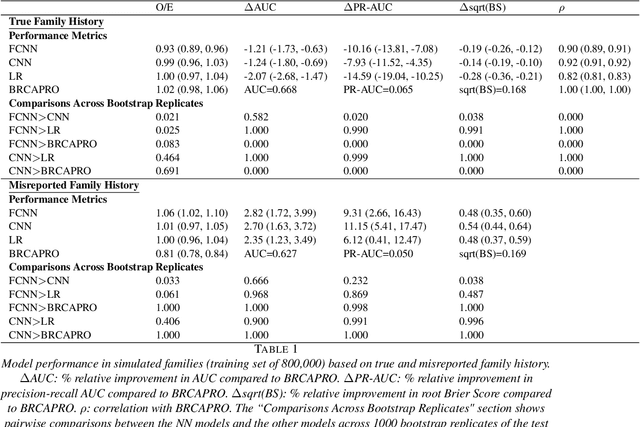
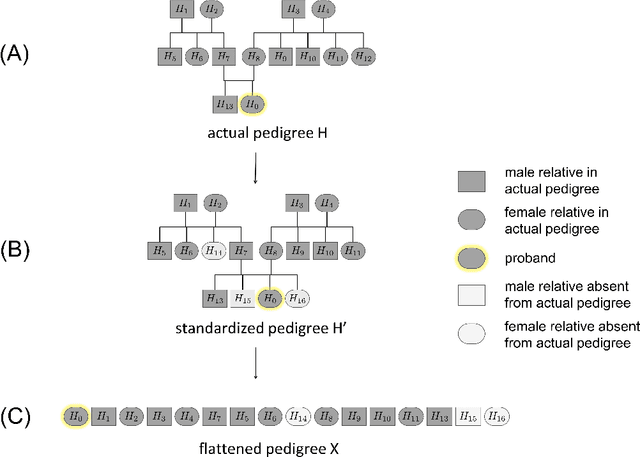
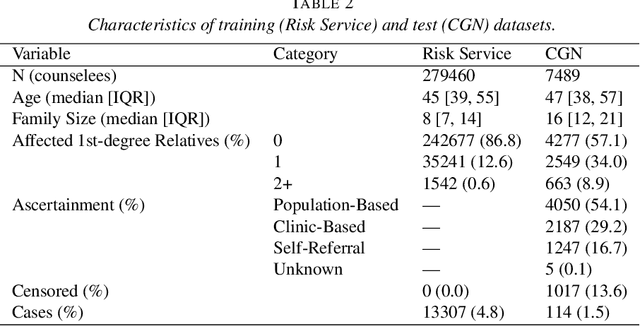
Family history is a major risk factor for many types of cancer. Mendelian risk prediction models translate family histories into cancer risk predictions based on knowledge of cancer susceptibility genes. These models are widely used in clinical practice to help identify high-risk individuals. Mendelian models leverage the entire family history, but they rely on many assumptions about cancer susceptibility genes that are either unrealistic or challenging to validate due to low mutation prevalence. Training more flexible models, such as neural networks, on large databases of pedigrees can potentially lead to accuracy gains. In this paper, we develop a framework to apply neural networks to family history data and investigate their ability to learn inherited susceptibility to cancer. While there is an extensive literature on neural networks and their state-of-the-art performance in many tasks, there is little work applying them to family history data. We propose adaptations of fully-connected neural networks and convolutional neural networks to pedigrees. In data simulated under Mendelian inheritance, we demonstrate that our proposed neural network models are able to achieve nearly optimal prediction performance. Moreover, when the observed family history includes misreported cancer diagnoses, neural networks are able to outperform the Mendelian BRCAPRO model embedding the correct inheritance laws. Using a large dataset of over 200,000 family histories, the Risk Service cohort, we train prediction models for future risk of breast cancer. We validate the models using data from the Cancer Genetics Network.
Robust Lineage Reconstruction from High-Dimensional Single-Cell Data
Jan 12, 2016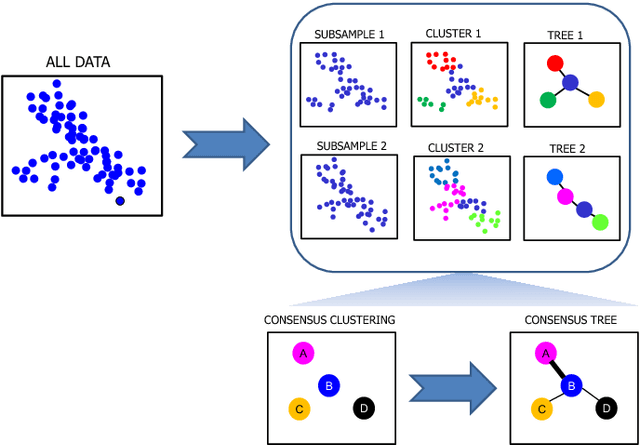
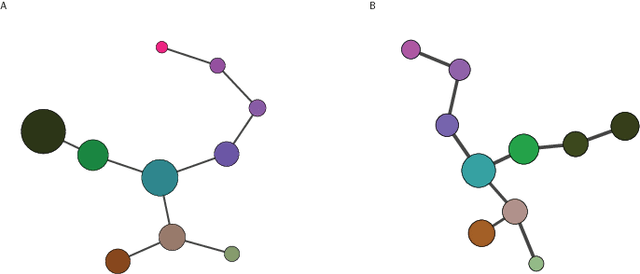
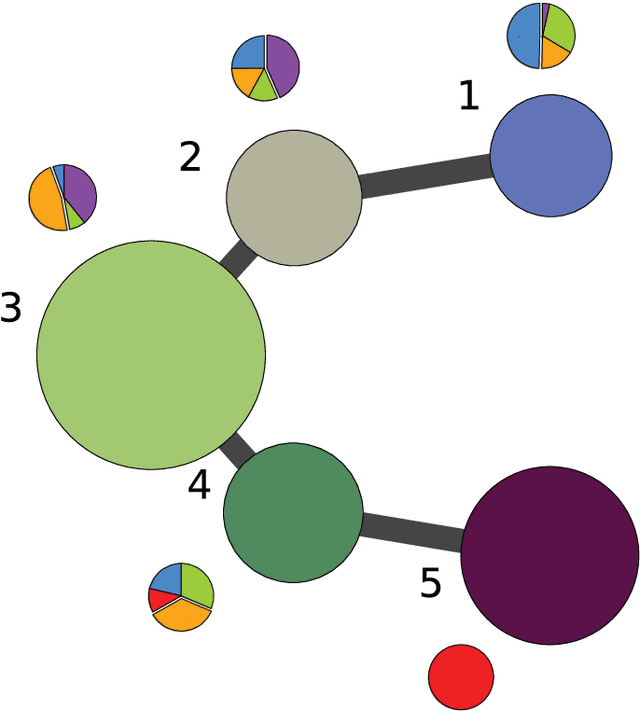
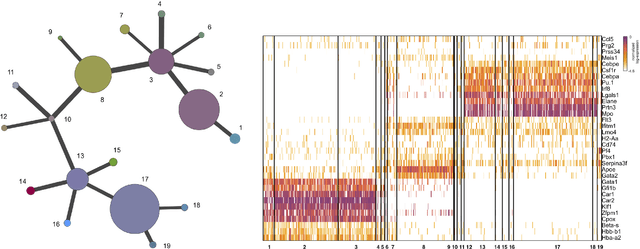
Single-cell gene expression data provide invaluable resources for systematic characterization of cellular hierarchy in multi-cellular organisms. However, cell lineage reconstruction is still often associated with significant uncertainty due to technological constraints. Such uncertainties have not been taken into account in current methods. We present ECLAIR, a novel computational method for the statistical inference of cell lineage relationships from single-cell gene expression data. ECLAIR uses an ensemble approach to improve the robustness of lineage predictions, and provides a quantitative estimate of the uncertainty of lineage branchings. We show that the application of ECLAIR to published datasets successfully reconstructs known lineage relationships and significantly improves the robustness of predictions. In conclusion, ECLAIR is a powerful bioinformatics tool for single-cell data analysis. It can be used for robust lineage reconstruction with quantitative estimate of prediction accuracy.