 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeSigmaDock: Untwisting Molecular Docking With Fragment-Based SE(3) Diffusion
Nov 06, 2025Determining the binding pose of a ligand to a protein, known as molecular docking, is a fundamental task in drug discovery. Generative approaches promise faster, improved, and more diverse pose sampling than physics-based methods, but are often hindered by chemically implausible outputs, poor generalisability, and high computational cost. To address these challenges, we introduce a novel fragmentation scheme, leveraging inductive biases from structural chemistry, to decompose ligands into rigid-body fragments. Building on this decomposition, we present SigmaDock, an SE(3) Riemannian diffusion model that generates poses by learning to reassemble these rigid bodies within the binding pocket. By operating at the level of fragments in SE(3), SigmaDock exploits well-established geometric priors while avoiding overly complex diffusion processes and unstable training dynamics. Experimentally, we show SigmaDock achieves state-of-the-art performance, reaching Top-1 success rates (RMSD<2 & PB-valid) above 79.9% on the PoseBusters set, compared to 12.7-30.8% reported by recent deep learning approaches, whilst demonstrating consistent generalisation to unseen proteins. SigmaDock is the first deep learning approach to surpass classical physics-based docking under the PB train-test split, marking a significant leap forward in the reliability and feasibility of deep learning for molecular modelling.
Assessing the Chemical Intelligence of Large Language Models
May 12, 2025Large Language Models are versatile, general-purpose tools with a wide range of applications. Recently, the advent of "reasoning models" has led to substantial improvements in their abilities in advanced problem-solving domains such as mathematics and software engineering. In this work, we assessed the ability of reasoning models to directly perform chemistry tasks, without any assistance from external tools. We created a novel benchmark, called ChemIQ, which consists of 796 questions assessing core concepts in organic chemistry, focused on molecular comprehension and chemical reasoning. Unlike previous benchmarks, which primarily use multiple choice formats, our approach requires models to construct short-answer responses, more closely reflecting real-world applications. The reasoning models, exemplified by OpenAI's o3-mini, correctly answered 28%-59% of questions depending on the reasoning level used, with higher reasoning levels significantly increasing performance on all tasks. These models substantially outperformed the non-reasoning model, GPT-4o, which achieved only 7% accuracy. We found that Large Language Models can now convert SMILES strings to IUPAC names, a task earlier models were unable to perform. Additionally, we show that the latest reasoning models can elucidate structures from 1H and 13C NMR data, correctly generating SMILES strings for 74% of molecules containing up to 10 heavy atoms, and in one case solving a structure comprising 21 heavy atoms. For each task, we found evidence that the reasoning process mirrors that of a human chemist. Our results demonstrate that the latest reasoning models have the ability to perform advanced chemical reasoning.
Transformers trained on proteins can learn to attend to Euclidean distance
Feb 03, 2025While conventional Transformers generally operate on sequence data, they can be used in conjunction with structure models, typically SE(3)-invariant or equivariant graph neural networks (GNNs), for 3D applications such as protein structure modelling. These hybrids typically involve either (1) preprocessing/tokenizing structural features as input for Transformers or (2) taking Transformer embeddings and processing them within a structural representation. However, there is evidence that Transformers can learn to process structural information on their own, such as the AlphaFold3 structural diffusion model. In this work we show that Transformers can function independently as structure models when passed linear embeddings of coordinates. We first provide a theoretical explanation for how Transformers can learn to filter attention as a 3D Gaussian with learned variance. We then validate this theory using both simulated 3D points and in the context of masked token prediction for proteins. Finally, we show that pre-training protein Transformer encoders with structure improves performance on a downstream task, yielding better performance than custom structural models. Together, this work provides a basis for using standard Transformers as hybrid structure-language models.
Context-Guided Diffusion for Out-of-Distribution Molecular and Protein Design
Jul 16, 2024
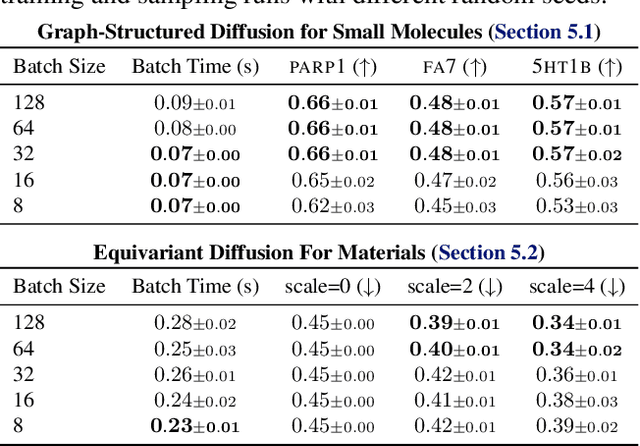


Generative models have the potential to accelerate key steps in the discovery of novel molecular therapeutics and materials. Diffusion models have recently emerged as a powerful approach, excelling at unconditional sample generation and, with data-driven guidance, conditional generation within their training domain. Reliably sampling from high-value regions beyond the training data, however, remains an open challenge -- with current methods predominantly focusing on modifying the diffusion process itself. In this paper, we develop context-guided diffusion (CGD), a simple plug-and-play method that leverages unlabeled data and smoothness constraints to improve the out-of-distribution generalization of guided diffusion models. We demonstrate that this approach leads to substantial performance gains across various settings, including continuous, discrete, and graph-structured diffusion processes with applications across drug discovery, materials science, and protein design.
ABodyBuilder3: Improved and scalable antibody structure predictions
May 31, 2024Accurate prediction of antibody structure is a central task in the design and development of monoclonal antibodies, notably to understand both their developability and their binding properties. In this article, we introduce ABodyBuilder3, an improved and scalable antibody structure prediction model based on ImmuneBuilder. We achieve a new state-of-the-art accuracy in the modelling of CDR loops by leveraging language model embeddings, and show how predicted structures can be further improved through careful relaxation strategies. Finally, we incorporate a predicted Local Distance Difference Test into the model output to allow for a more accurate estimation of uncertainties.
De novo antibody design with SE diffusion
May 13, 2024We introduce IgDiff, an antibody variable domain diffusion model based on a general protein backbone diffusion framework which was extended to handle multiple chains. Assessing the designability and novelty of the structures generated with our model, we find that IgDiff produces highly designable antibodies that can contain novel binding regions. The backbone dihedral angles of sampled structures show good agreement with a reference antibody distribution. We verify these designed antibodies experimentally and find that all express with high yield. Finally, we compare our model with a state-of-the-art generative backbone diffusion model on a range of antibody design tasks, such as the design of the complementarity determining regions or the pairing of a light chain to an existing heavy chain, and show improved properties and designability.
Large scale paired antibody language models
Mar 26, 2024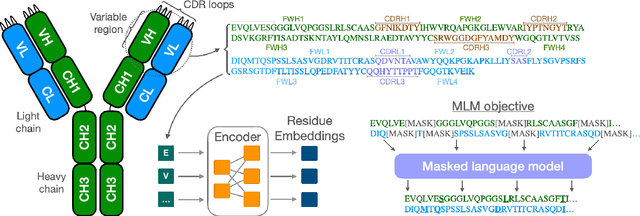
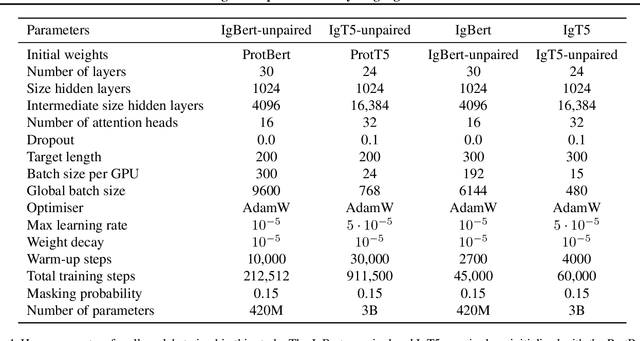
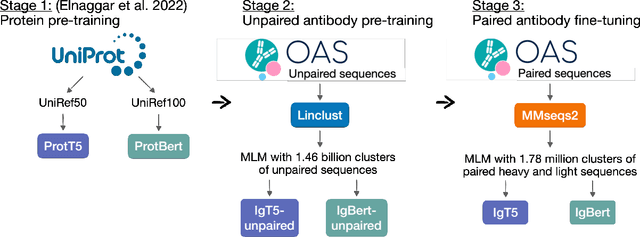
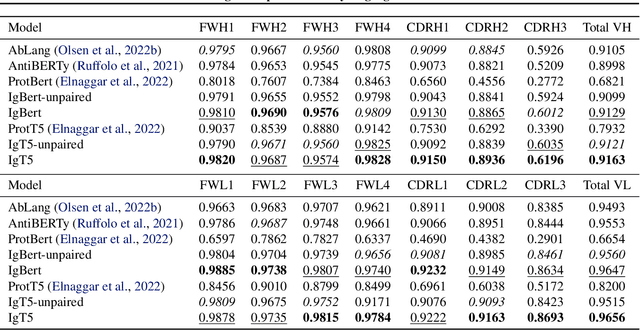
Antibodies are proteins produced by the immune system that can identify and neutralise a wide variety of antigens with high specificity and affinity, and constitute the most successful class of biotherapeutics. With the advent of next-generation sequencing, billions of antibody sequences have been collected in recent years, though their application in the design of better therapeutics has been constrained by the sheer volume and complexity of the data. To address this challenge, we present IgBert and IgT5, the best performing antibody-specific language models developed to date which can consistently handle both paired and unpaired variable region sequences as input. These models are trained comprehensively using the more than two billion unpaired sequences and two million paired sequences of light and heavy chains present in the Observed Antibody Space dataset. We show that our models outperform existing antibody and protein language models on a diverse range of design and regression tasks relevant to antibody engineering. This advancement marks a significant leap forward in leveraging machine learning, large scale data sets and high-performance computing for enhancing antibody design for therapeutic development.
Inverse folding for antibody sequence design using deep learning
Oct 30, 2023We consider the problem of antibody sequence design given 3D structural information. Building on previous work, we propose a fine-tuned inverse folding model that is specifically optimised for antibody structures and outperforms generic protein models on sequence recovery and structure robustness when applied on antibodies, with notable improvement on the hypervariable CDR-H3 loop. We study the canonical conformations of complementarity-determining regions and find improved encoding of these loops into known clusters. Finally, we consider the applications of our model to drug discovery and binder design and evaluate the quality of proposed sequences using physics-based methods.
Ranking of Communities in Multiplex Spatiotemporal Models of Brain Dynamics
Mar 17, 2022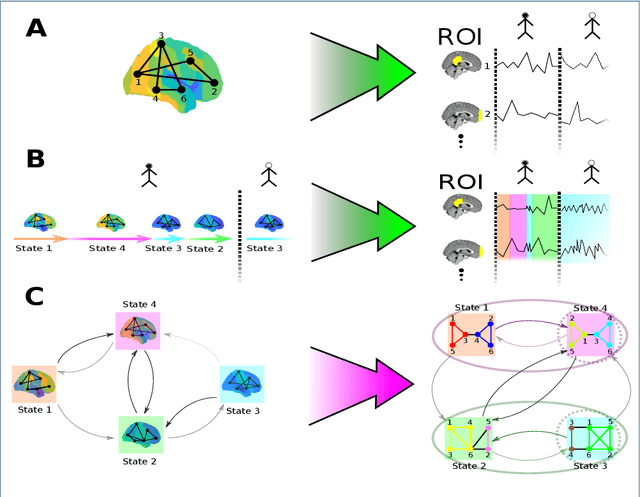

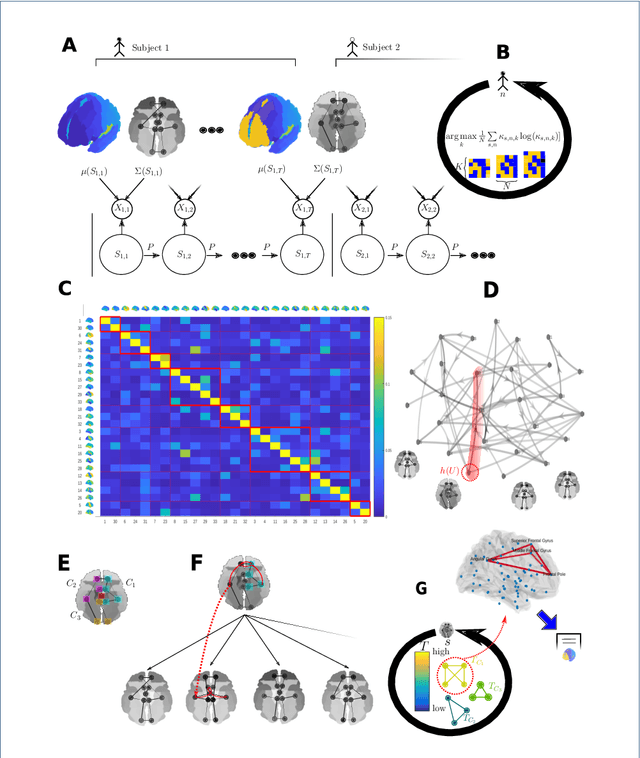
As a relatively new field, network neuroscience has tended to focus on aggregate behaviours of the brain averaged over many successive experiments or over long recordings in order to construct robust brain models. These models are limited in their ability to explain dynamic state changes in the brain which occurs spontaneously as a result of normal brain function. Hidden Markov Models (HMMs) trained on neuroimaging time series data have since arisen as a method to produce dynamical models that are easy to train but can be difficult to fully parametrise or analyse. We propose an interpretation of these neural HMMs as multiplex brain state graph models we term Hidden Markov Graph Models (HMGMs). This interpretation allows for dynamic brain activity to be analysed using the full repertoire of network analysis techniques. Furthermore, we propose a general method for selecting HMM hyperparameters in the absence of external data, based on the principle of maximum entropy, and use this to select the number of layers in the multiplex model. We produce a new tool for determining important communities of brain regions using a spatiotemporal random walk-based procedure that takes advantage of the underlying Markov structure of the model. Our analysis of real multi-subject fMRI data provides new results that corroborate the modular processing hypothesis of the brain at rest as well as contributing new evidence of functional overlap between and within dynamic brain state communities. Our analysis pipeline provides a way to characterise dynamic network activity of the brain under novel behaviours or conditions.
* Part of the Special Issue on Community Structure in Networks 2021 (35 Pages, first 22 for main text)
The prospects of quantum computing in computational molecular biology
May 26, 2020
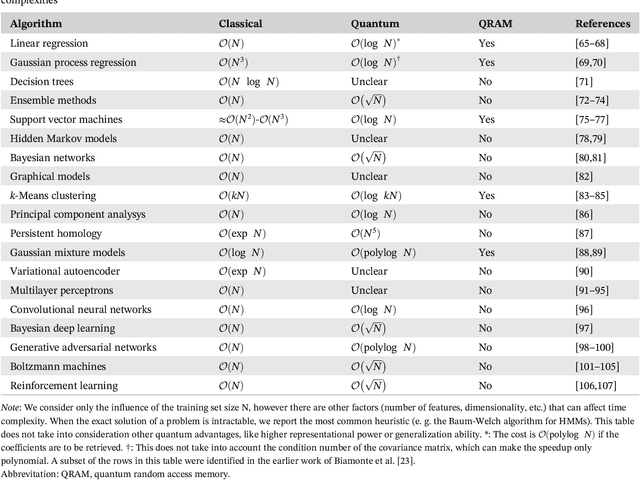
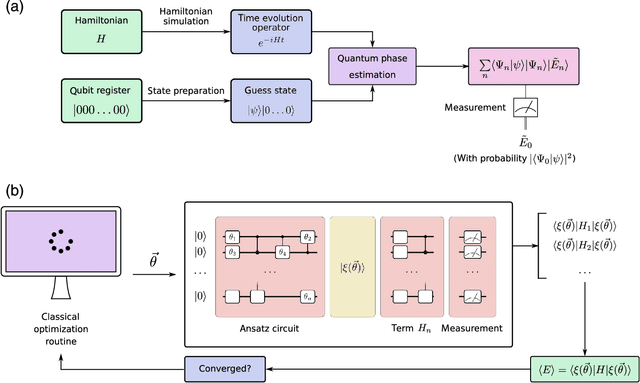

Quantum computers can in principle solve certain problems exponentially more quickly than their classical counterparts. We have not yet reached the advent of useful quantum computation, but when we do, it will affect nearly all scientific disciplines. In this review, we examine how current quantum algorithms could revolutionize computational biology and bioinformatics. There are potential benefits across the entire field, from the ability to process vast amounts of information and run machine learning algorithms far more efficiently, to algorithms for quantum simulation that are poised to improve computational calculations in drug discovery, to quantum algorithms for optimization that may advance fields from protein structure prediction to network analysis. However, these exciting prospects are susceptible to "hype", and it is also important to recognize the caveats and challenges in this new technology. Our aim is to introduce the promise and limitations of emerging quantum computing technologies in the areas of computational molecular biology and bioinformatics.
* 23 pages, 3 figures