 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeTransformers trained on proteins can learn to attend to Euclidean distance
Feb 03, 2025While conventional Transformers generally operate on sequence data, they can be used in conjunction with structure models, typically SE(3)-invariant or equivariant graph neural networks (GNNs), for 3D applications such as protein structure modelling. These hybrids typically involve either (1) preprocessing/tokenizing structural features as input for Transformers or (2) taking Transformer embeddings and processing them within a structural representation. However, there is evidence that Transformers can learn to process structural information on their own, such as the AlphaFold3 structural diffusion model. In this work we show that Transformers can function independently as structure models when passed linear embeddings of coordinates. We first provide a theoretical explanation for how Transformers can learn to filter attention as a 3D Gaussian with learned variance. We then validate this theory using both simulated 3D points and in the context of masked token prediction for proteins. Finally, we show that pre-training protein Transformer encoders with structure improves performance on a downstream task, yielding better performance than custom structural models. Together, this work provides a basis for using standard Transformers as hybrid structure-language models.
Assessing interaction recovery of predicted protein-ligand poses
Sep 30, 2024The field of protein-ligand pose prediction has seen significant advances in recent years, with machine learning-based methods now being commonly used in lieu of classical docking methods or even to predict all-atom protein-ligand complex structures. Most contemporary studies focus on the accuracy and physical plausibility of ligand placement to determine pose quality, often neglecting a direct assessment of the interactions observed with the protein. In this work, we demonstrate that ignoring protein-ligand interaction fingerprints can lead to overestimation of model performance, most notably in recent protein-ligand cofolding models which often fail to recapitulate key interactions.
De novo antibody design with SE diffusion
May 13, 2024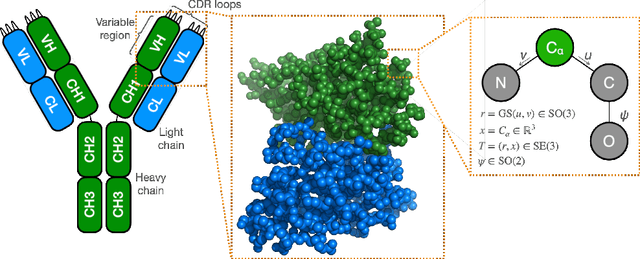



We introduce IgDiff, an antibody variable domain diffusion model based on a general protein backbone diffusion framework which was extended to handle multiple chains. Assessing the designability and novelty of the structures generated with our model, we find that IgDiff produces highly designable antibodies that can contain novel binding regions. The backbone dihedral angles of sampled structures show good agreement with a reference antibody distribution. We verify these designed antibodies experimentally and find that all express with high yield. Finally, we compare our model with a state-of-the-art generative backbone diffusion model on a range of antibody design tasks, such as the design of the complementarity determining regions or the pairing of a light chain to an existing heavy chain, and show improved properties and designability.
Inverse folding for antibody sequence design using deep learning
Oct 30, 2023We consider the problem of antibody sequence design given 3D structural information. Building on previous work, we propose a fine-tuned inverse folding model that is specifically optimised for antibody structures and outperforms generic protein models on sequence recovery and structure robustness when applied on antibodies, with notable improvement on the hypervariable CDR-H3 loop. We study the canonical conformations of complementarity-determining regions and find improved encoding of these loops into known clusters. Finally, we consider the applications of our model to drug discovery and binder design and evaluate the quality of proposed sequences using physics-based methods.