 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeRethinking Molecular Text Representations for LLMs: An Empirical Study
Jun 02, 2026Large language models (LLMs) are increasingly used for molecular tasks, but it remains unclear which molecular representation to use. We present a systematic benchmark evaluating LLM molecular competence across nine representations and eight chemical tasks. We benchmark 16 LLMs across five model families, including reasoning and non-reasoning variants, chemistry-specialized LLMs, and closed frontier models. Performance is strongly representation-dependent and no single representation wins across tasks, though CML is the best, followed by MolJSON, InChI, and then canonical SMILES. Explicit structured text representations (CML and MolJSON) dominate structural tasks; IUPAC dominates semantic tasks, winning molecule retrieval for all 16 LLMs; and SMILES variants are rarely optimal despite their prevalence in pretraining. Chemistry-specialized models perform well with SMILES at the cost of large degradations with structured text representations, suggesting SMILES-only evaluation rewards specialization that does not generalize. Using LLM-as-a-judge, we find that IUPAC produces the highest fraction of correct molecule generations. A mechanistic study via tokenization audits, linear probes and attention shows that representations are encoded differently inside the model; for example, structured representations require higher attention across the molecular span. Our results argue against representation-invariant evaluation and motivate task-aware representation routing for LLM-based chemistry.
On Improving Graph Neural Networks for QSAR by Pre-training on Extended-Connectivity Fingerprints
May 11, 2026Molecular Graph Neural Networks (GNNs) are increasingly common in drug discovery, particularly for Quantitative Structure-Activity Relationship (QSAR) studies; yet, their superiority compared to classical molecular featurisation approaches is disputed. We report a general strategy for improving GNNs for QSAR by pre-training to predict Extended-Connectivity Fingerprints (ECFP). We validate our approach with statistical tests and challenging out-of-distribution (OOD) splits. Across five out of six Biogen benchmarks, we observed a statistically significant improvement in standard performance metrics over all evaluated baselines when using ECFP pre-trained GNNs. However, for more heterogeneous datasets and more complex endpoints, such as binding affinity prediction, pre-trained GNNs underperformed in OOD settings. Importantly, we investigated the impact of substructure-level data leakage during pre-training on downstream performance. While we identified scenarios where pre-training on ECFPs was less effective, our findings show that ECFP-based pre-training can enhance downstream OOD performance on a diverse set of practically relevant QSAR tasks.
SigmaDock: Untwisting Molecular Docking With Fragment-Based SE(3) Diffusion
Nov 06, 2025Determining the binding pose of a ligand to a protein, known as molecular docking, is a fundamental task in drug discovery. Generative approaches promise faster, improved, and more diverse pose sampling than physics-based methods, but are often hindered by chemically implausible outputs, poor generalisability, and high computational cost. To address these challenges, we introduce a novel fragmentation scheme, leveraging inductive biases from structural chemistry, to decompose ligands into rigid-body fragments. Building on this decomposition, we present SigmaDock, an SE(3) Riemannian diffusion model that generates poses by learning to reassemble these rigid bodies within the binding pocket. By operating at the level of fragments in SE(3), SigmaDock exploits well-established geometric priors while avoiding overly complex diffusion processes and unstable training dynamics. Experimentally, we show SigmaDock achieves state-of-the-art performance, reaching Top-1 success rates (RMSD<2 & PB-valid) above 79.9% on the PoseBusters set, compared to 12.7-30.8% reported by recent deep learning approaches, whilst demonstrating consistent generalisation to unseen proteins. SigmaDock is the first deep learning approach to surpass classical physics-based docking under the PB train-test split, marking a significant leap forward in the reliability and feasibility of deep learning for molecular modelling.
Context-Guided Diffusion for Out-of-Distribution Molecular and Protein Design
Jul 16, 2024
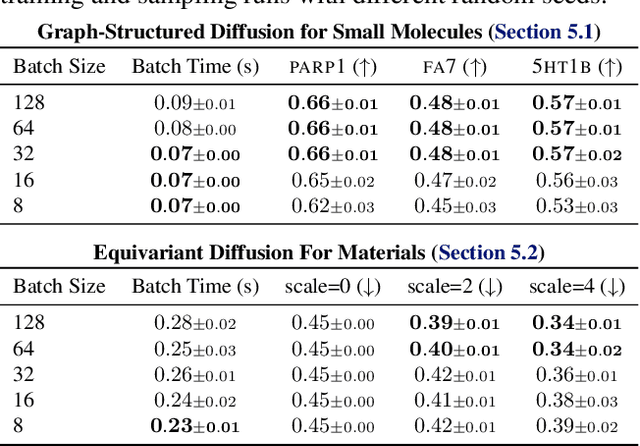


Generative models have the potential to accelerate key steps in the discovery of novel molecular therapeutics and materials. Diffusion models have recently emerged as a powerful approach, excelling at unconditional sample generation and, with data-driven guidance, conditional generation within their training domain. Reliably sampling from high-value regions beyond the training data, however, remains an open challenge -- with current methods predominantly focusing on modifying the diffusion process itself. In this paper, we develop context-guided diffusion (CGD), a simple plug-and-play method that leverages unlabeled data and smoothness constraints to improve the out-of-distribution generalization of guided diffusion models. We demonstrate that this approach leads to substantial performance gains across various settings, including continuous, discrete, and graph-structured diffusion processes with applications across drug discovery, materials science, and protein design.
Drug Discovery under Covariate Shift with Domain-Informed Prior Distributions over Functions
Jul 14, 2023



Accelerating the discovery of novel and more effective therapeutics is an important pharmaceutical problem in which deep learning is playing an increasingly significant role. However, real-world drug discovery tasks are often characterized by a scarcity of labeled data and significant covariate shift$\unicode{x2013}\unicode{x2013}$a setting that poses a challenge to standard deep learning methods. In this paper, we present Q-SAVI, a probabilistic model able to address these challenges by encoding explicit prior knowledge of the data-generating process into a prior distribution over functions, presenting researchers with a transparent and probabilistically principled way to encode data-driven modeling preferences. Building on a novel, gold-standard bioactivity dataset that facilitates a meaningful comparison of models in an extrapolative regime, we explore different approaches to induce data shift and construct a challenging evaluation setup. We then demonstrate that using Q-SAVI to integrate contextualized prior knowledge of drug-like chemical space into the modeling process affords substantial gains in predictive accuracy and calibration, outperforming a broad range of state-of-the-art self-supervised pre-training and domain adaptation techniques.
Exploring QSAR Models for Activity-Cliff Prediction
Jan 31, 2023Pairs of similar compounds that only differ by a small structural modification but exhibit a large difference in their binding affinity for a given target are known as activity cliffs (ACs). It has been hypothesised that quantitative structure-activity relationship (QSAR) models struggle to predict ACs and that ACs thus form a major source of prediction error. However, a study to explore the AC-prediction power of modern QSAR methods and its relationship to general QSAR-prediction performance is lacking. We systematically construct nine distinct QSAR models by combining three molecular representation methods (extended-connectivity fingerprints, physicochemical-descriptor vectors and graph isomorphism networks) with three regression techniques (random forests, k-nearest neighbours and multilayer perceptrons); we then use each resulting model to classify pairs of similar compounds as ACs or non-ACs and to predict the activities of individual molecules in three case studies: dopamine receptor D2, factor Xa, and SARS-CoV-2 main protease. We observe low AC-sensitivity amongst the tested models when the activities of both compounds are unknown, but a substantial increase in AC-sensitivity when the actual activity of one of the compounds is given. Graph isomorphism features are found to be competitive with or superior to classical molecular representations for AC-classification and can thus be employed as baseline AC-prediction models or simple compound-optimisation tools. For general QSAR-prediction, however, extended-connectivity fingerprints still consistently deliver the best performance. Our results provide strong support for the hypothesis that indeed QSAR methods frequently fail to predict ACs. We propose twin-network training for deep learning models as a potential future pathway to increase AC-sensitivity and thus overall QSAR performance.
The prospects of quantum computing in computational molecular biology
May 26, 2020
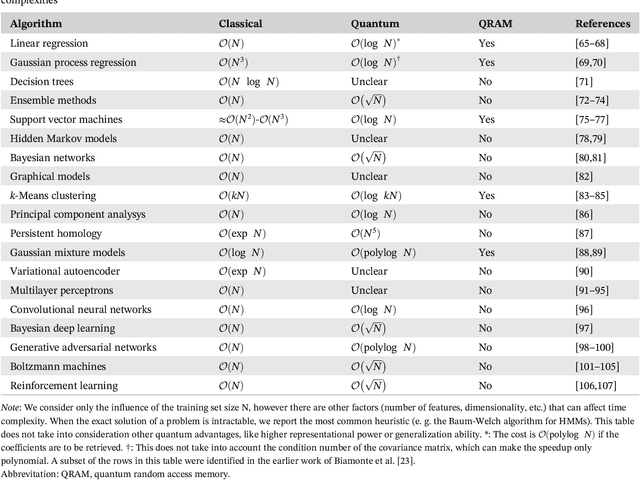
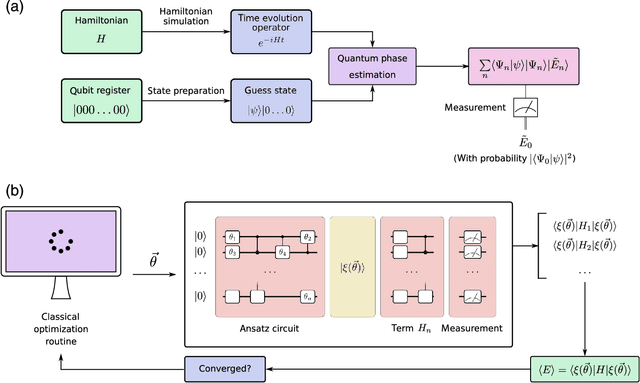

Quantum computers can in principle solve certain problems exponentially more quickly than their classical counterparts. We have not yet reached the advent of useful quantum computation, but when we do, it will affect nearly all scientific disciplines. In this review, we examine how current quantum algorithms could revolutionize computational biology and bioinformatics. There are potential benefits across the entire field, from the ability to process vast amounts of information and run machine learning algorithms far more efficiently, to algorithms for quantum simulation that are poised to improve computational calculations in drug discovery, to quantum algorithms for optimization that may advance fields from protein structure prediction to network analysis. However, these exciting prospects are susceptible to "hype", and it is also important to recognize the caveats and challenges in this new technology. Our aim is to introduce the promise and limitations of emerging quantum computing technologies in the areas of computational molecular biology and bioinformatics.
* 23 pages, 3 figures