 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeDrug Discovery under Covariate Shift with Domain-Informed Prior Distributions over Functions
Jul 14, 2023



Accelerating the discovery of novel and more effective therapeutics is an important pharmaceutical problem in which deep learning is playing an increasingly significant role. However, real-world drug discovery tasks are often characterized by a scarcity of labeled data and significant covariate shift$\unicode{x2013}\unicode{x2013}$a setting that poses a challenge to standard deep learning methods. In this paper, we present Q-SAVI, a probabilistic model able to address these challenges by encoding explicit prior knowledge of the data-generating process into a prior distribution over functions, presenting researchers with a transparent and probabilistically principled way to encode data-driven modeling preferences. Building on a novel, gold-standard bioactivity dataset that facilitates a meaningful comparison of models in an extrapolative regime, we explore different approaches to induce data shift and construct a challenging evaluation setup. We then demonstrate that using Q-SAVI to integrate contextualized prior knowledge of drug-like chemical space into the modeling process affords substantial gains in predictive accuracy and calibration, outperforming a broad range of state-of-the-art self-supervised pre-training and domain adaptation techniques.
Benchmarking Accuracy and Generalizability of Four Graph Neural Networks Using Large In Vitro ADME Datasets from Different Chemical Spaces
Nov 27, 2021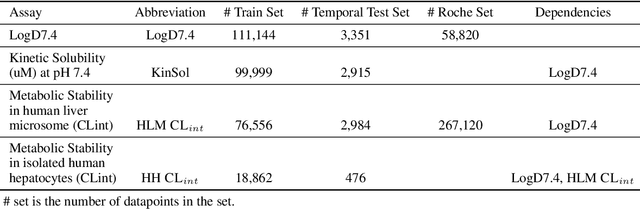
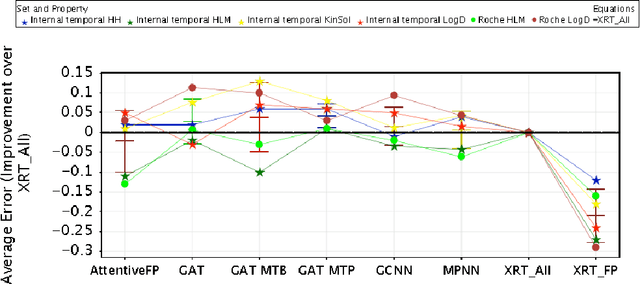

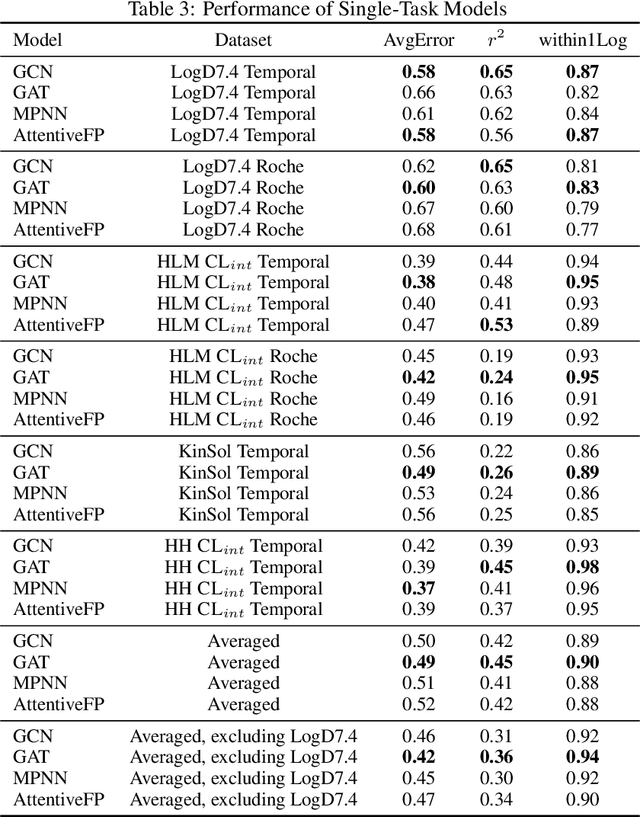
In this work, we benchmark a variety of single- and multi-task graph neural network (GNN) models against lower-bar and higher-bar traditional machine learning approaches employing human engineered molecular features. We consider four GNN variants -- Graph Convolutional Network (GCN), Graph Attention Network (GAT), Message Passing Neural Network (MPNN), and Attentive Fingerprint (AttentiveFP). So far deep learning models have been primarily benchmarked using lower-bar traditional models solely based on fingerprints, while more realistic benchmarks employing fingerprints, whole-molecule descriptors and predictions from other related endpoints (e.g., LogD7.4) appear to be scarce for industrial ADME datasets. In addition to time-split test sets based on Genentech data, this study benefits from the availability of measurements from an external chemical space (Roche data). We identify GAT as a promising approach to implementing deep learning models. While all GNN models significantly outperform lower-bar benchmark traditional models solely based on fingerprints, only GATs seem to offer a small but consistent improvement over higher-bar benchmark traditional models. Finally, the accuracy of in vitro assays from different laboratories predicting the same experimental endpoints appears to be comparable with the accuracy of GAT single-task models, suggesting that most of the observed error from the models is a function of the experimental error propagation.