 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMake Literature-Based Discovery Great Again through Reproducible Pipelines
Feb 23, 2025By connecting disparate sources of scientific literature, literature\-/based discovery (LBD) methods help to uncover new knowledge and generate new research hypotheses that cannot be found from domain-specific documents alone. Our work focuses on bisociative LBD methods that combine bisociative reasoning with LBD techniques. The paper presents LBD through the lens of reproducible science to ensure the reproducibility of LBD experiments, overcome the inconsistent use of benchmark datasets and methods, trigger collaboration, and advance the LBD field toward more robust and impactful scientific discoveries. The main novelty of this study is a collection of Jupyter Notebooks that illustrate the steps of the bisociative LBD process, including data acquisition, text preprocessing, hypothesis formulation, and evaluation. The contributed notebooks implement a selection of traditional LBD approaches, as well as our own ensemble-based, outlier-based, and link prediction-based approaches. The reader can benefit from hands-on experience with LBD through open access to benchmark datasets, code reuse, and a ready-to-run Docker recipe that ensures reproducibility of the selected LBD methods.
AHAM: Adapt, Help, Ask, Model -- Harvesting LLMs for literature mining
Dec 25, 2023In an era marked by a rapid increase in scientific publications, researchers grapple with the challenge of keeping pace with field-specific advances. We present the `AHAM' methodology and a metric that guides the domain-specific \textbf{adapt}ation of the BERTopic topic modeling framework to improve scientific text analysis. By utilizing the LLaMa2 generative language model, we generate topic definitions via one-shot learning by crafting prompts with the \textbf{help} of domain experts to guide the LLM for literature mining by \textbf{asking} it to model the topic names. For inter-topic similarity evaluation, we leverage metrics from language generation and translation processes to assess lexical and semantic similarity of the generated topics. Our system aims to reduce both the ratio of outlier topics to the total number of topics and the similarity between topic definitions. The methodology has been assessed on a newly gathered corpus of scientific papers on literature-based discovery. Through rigorous evaluation by domain experts, AHAM has been validated as effective in uncovering intriguing and novel insights within broad research areas. We explore the impact of domain adaptation of sentence-transformers for the task of topic \textbf{model}ing using two datasets, each specialized to specific scientific domains within arXiv and medarxiv. We evaluate the impact of data size, the niche of adaptation, and the importance of domain adaptation. Our results suggest a strong interaction between domain adaptation and topic modeling precision in terms of outliers and topic definitions.
Drug Repurposing for COVID-19 via Knowledge Graph Completion
Oct 19, 2020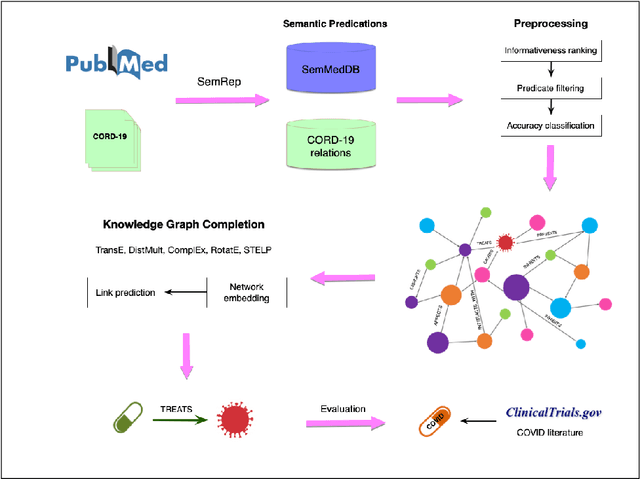
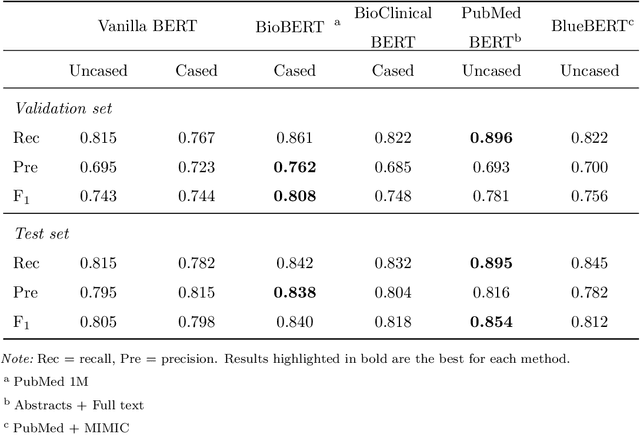
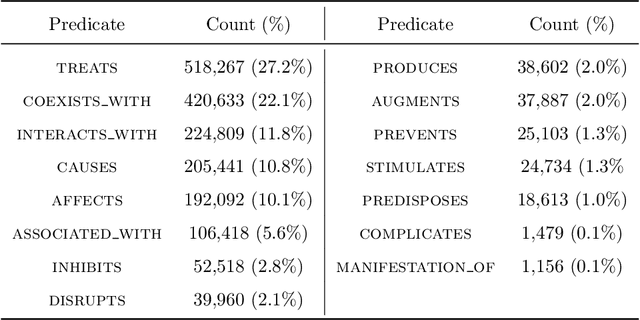
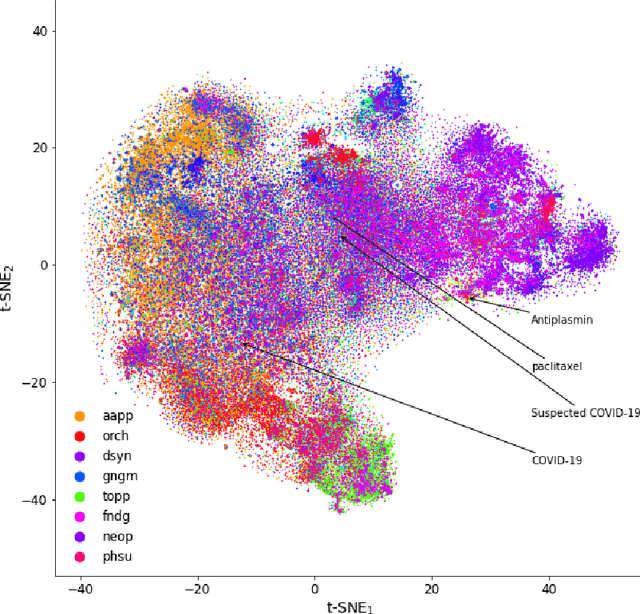
Objective: To discover candidate drugs to repurpose for COVID-19 using literature-derived knowledge and knowledge graph completion methods. Methods: We propose a novel, integrative, and neural network-based literature-based discovery (LBD) approach to identify drug candidates from both PubMed and COVID-19-focused research literature. Our approach relies on semantic triples extracted using SemRep (via SemMedDB). We identified an informative subset of semantic triples using filtering rules and an accuracy classifier developed on a BERT variant, and used this subset to construct a knowledge graph. Five SOTA, neural knowledge graph completion algorithms were used to predict drug repurposing candidates. The models were trained and assessed using a time slicing approach and the predicted drugs were compared with a list of drugs reported in the literature and evaluated in clinical trials. These models were complemented by a discovery pattern-based approach. Results: Accuracy classifier based on PubMedBERT achieved the best performance (F1= 0.854) in classifying semantic predications. Among five knowledge graph completion models, TransE outperformed others (MR = 0.923, Hits@1=0.417). Some known drugs linked to COVID-19 in the literature were identified, as well as some candidate drugs that have not yet been studied. Discovery patterns enabled generation of plausible hypotheses regarding the relationships between the candidate drugs and COVID-19. Among them, five highly ranked and novel drugs (paclitaxel, SB 203580, alpha 2-antiplasmin, pyrrolidine dithiocarbamate, and butylated hydroxytoluene) with their mechanistic explanations were further discussed. Conclusion: We show that an LBD approach can be feasible for discovering drug candidates for COVID-19, and for generating mechanistic explanations. Our approach can be generalized to other diseases as well as to other clinical questions.
Rasch-based high-dimensionality data reduction and class prediction with applications to microarray gene expression data
Jun 05, 2010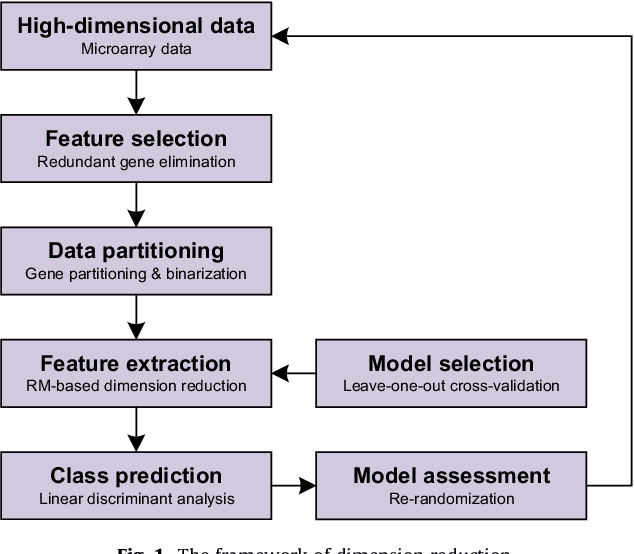
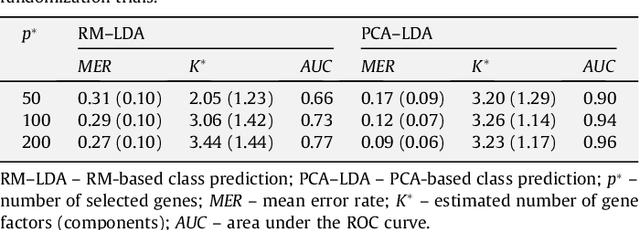
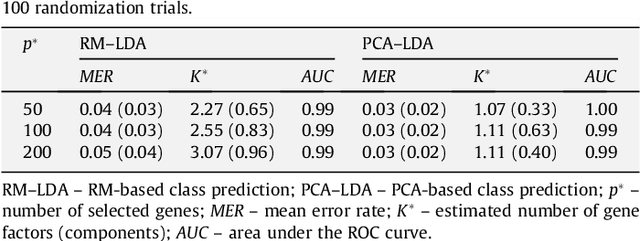
Class prediction is an important application of microarray gene expression data analysis. The high-dimensionality of microarray data, where number of genes (variables) is very large compared to the number of samples (obser- vations), makes the application of many prediction techniques (e.g., logistic regression, discriminant analysis) difficult. An efficient way to solve this prob- lem is by using dimension reduction statistical techniques. Increasingly used in psychology-related applications, Rasch model (RM) provides an appealing framework for handling high-dimensional microarray data. In this paper, we study the potential of RM-based modeling in dimensionality reduction with binarized microarray gene expression data and investigate its prediction ac- curacy in the context of class prediction using linear discriminant analysis. Two different publicly available microarray data sets are used to illustrate a general framework of the approach. Performance of the proposed method is assessed by re-randomization scheme using principal component analysis (PCA) as a benchmark method. Our results show that RM-based dimension reduction is as effective as PCA-based dimension reduction. The method is general and can be applied to the other high-dimensional data problems.
Chi-square-based scoring function for categorization of MEDLINE citations
Jun 05, 2010
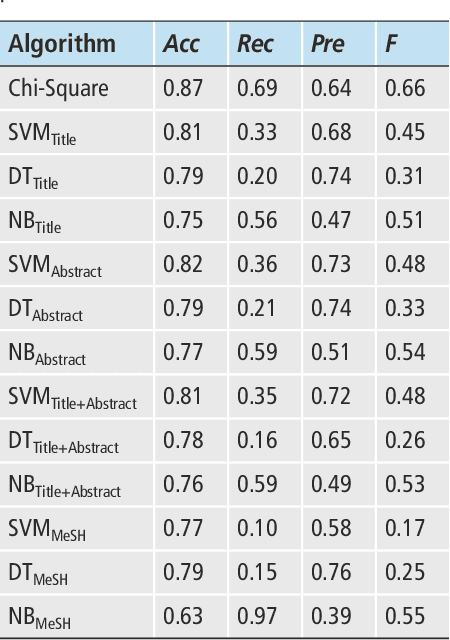
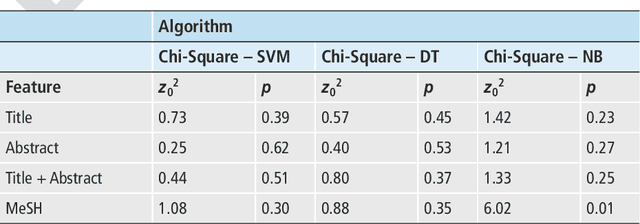
Objectives: Text categorization has been used in biomedical informatics for identifying documents containing relevant topics of interest. We developed a simple method that uses a chi-square-based scoring function to determine the likelihood of MEDLINE citations containing genetic relevant topic. Methods: Our procedure requires construction of a genetic and a nongenetic domain document corpus. We used MeSH descriptors assigned to MEDLINE citations for this categorization task. We compared frequencies of MeSH descriptors between two corpora applying chi-square test. A MeSH descriptor was considered to be a positive indicator if its relative observed frequency in the genetic domain corpus was greater than its relative observed frequency in the nongenetic domain corpus. The output of the proposed method is a list of scores for all the citations, with the highest score given to those citations containing MeSH descriptors typical for the genetic domain. Results: Validation was done on a set of 734 manually annotated MEDLINE citations. It achieved predictive accuracy of 0.87 with 0.69 recall and 0.64 precision. We evaluated the method by comparing it to three machine learning algorithms (support vector machines, decision trees, na\"ive Bayes). Although the differences were not statistically significantly different, results showed that our chi-square scoring performs as good as compared machine learning algorithms. Conclusions: We suggest that the chi-square scoring is an effective solution to help categorize MEDLINE citations. The algorithm is implemented in the BITOLA literature-based discovery support system as a preprocessor for gene symbol disambiguation process.
* 34 pages, 2 figures