 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeRasch-based high-dimensionality data reduction and class prediction with applications to microarray gene expression data
Jun 05, 2010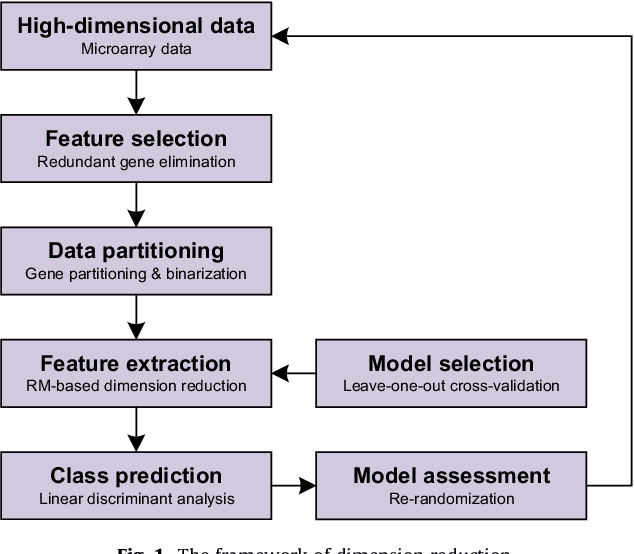
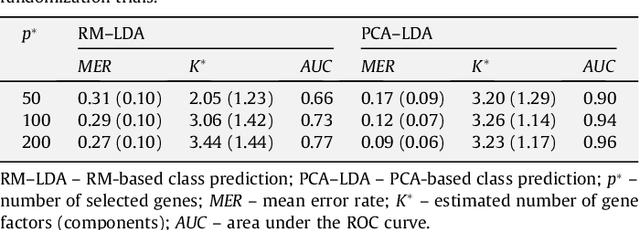
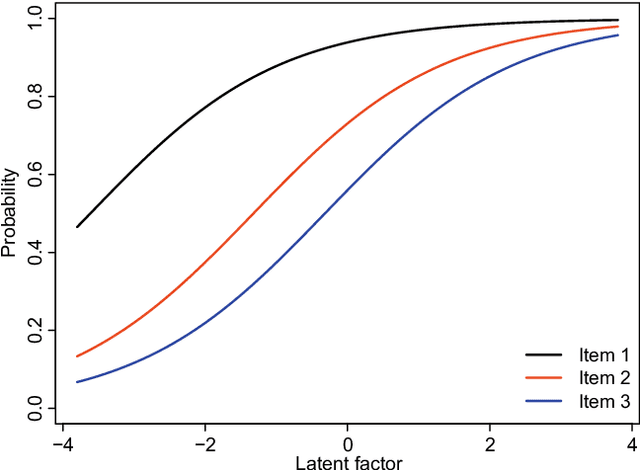
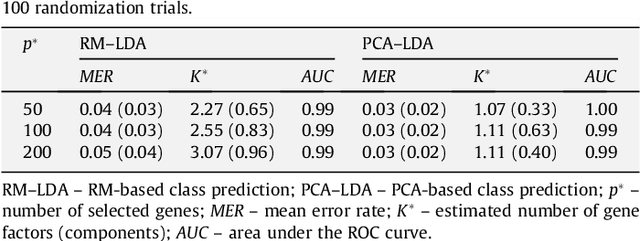
Class prediction is an important application of microarray gene expression data analysis. The high-dimensionality of microarray data, where number of genes (variables) is very large compared to the number of samples (obser- vations), makes the application of many prediction techniques (e.g., logistic regression, discriminant analysis) difficult. An efficient way to solve this prob- lem is by using dimension reduction statistical techniques. Increasingly used in psychology-related applications, Rasch model (RM) provides an appealing framework for handling high-dimensional microarray data. In this paper, we study the potential of RM-based modeling in dimensionality reduction with binarized microarray gene expression data and investigate its prediction ac- curacy in the context of class prediction using linear discriminant analysis. Two different publicly available microarray data sets are used to illustrate a general framework of the approach. Performance of the proposed method is assessed by re-randomization scheme using principal component analysis (PCA) as a benchmark method. Our results show that RM-based dimension reduction is as effective as PCA-based dimension reduction. The method is general and can be applied to the other high-dimensional data problems.
Chi-square-based scoring function for categorization of MEDLINE citations
Jun 05, 2010
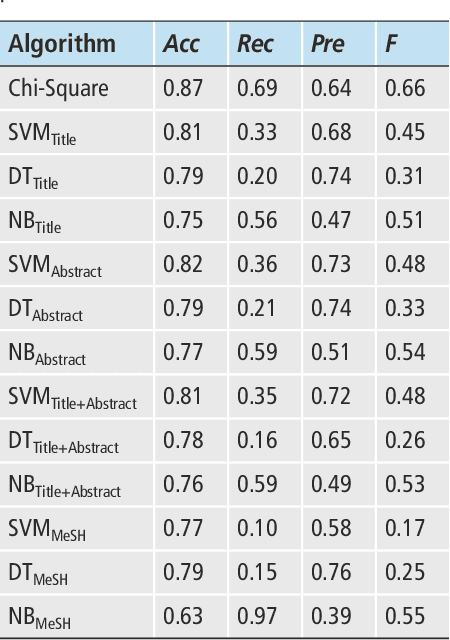
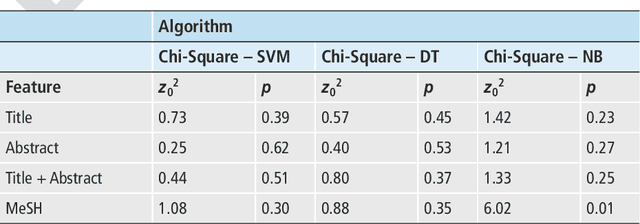
Objectives: Text categorization has been used in biomedical informatics for identifying documents containing relevant topics of interest. We developed a simple method that uses a chi-square-based scoring function to determine the likelihood of MEDLINE citations containing genetic relevant topic. Methods: Our procedure requires construction of a genetic and a nongenetic domain document corpus. We used MeSH descriptors assigned to MEDLINE citations for this categorization task. We compared frequencies of MeSH descriptors between two corpora applying chi-square test. A MeSH descriptor was considered to be a positive indicator if its relative observed frequency in the genetic domain corpus was greater than its relative observed frequency in the nongenetic domain corpus. The output of the proposed method is a list of scores for all the citations, with the highest score given to those citations containing MeSH descriptors typical for the genetic domain. Results: Validation was done on a set of 734 manually annotated MEDLINE citations. It achieved predictive accuracy of 0.87 with 0.69 recall and 0.64 precision. We evaluated the method by comparing it to three machine learning algorithms (support vector machines, decision trees, na\"ive Bayes). Although the differences were not statistically significantly different, results showed that our chi-square scoring performs as good as compared machine learning algorithms. Conclusions: We suggest that the chi-square scoring is an effective solution to help categorize MEDLINE citations. The algorithm is implemented in the BITOLA literature-based discovery support system as a preprocessor for gene symbol disambiguation process.
* 34 pages, 2 figures