 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeA Benchmark Dataset for Multimodal Prediction of Enzymatic Function Coupling DNA Sequences and Natural Language
Jul 21, 2024



Predicting gene function from its DNA sequence is a fundamental challenge in biology. Many deep learning models have been proposed to embed DNA sequences and predict their enzymatic function, leveraging information in public databases linking DNA sequences to an enzymatic function label. However, much of the scientific community's knowledge of biological function is not represented in these categorical labels, and is instead captured in unstructured text descriptions of mechanisms, reactions, and enzyme behavior. These descriptions are often captured alongside DNA sequences in biological databases, albeit in an unstructured manner. Deep learning of models predicting enzymatic function are likely to benefit from incorporating this multi-modal data encoding scientific knowledge of biological function. There is, however, no dataset designed for machine learning algorithms to leverage this multi-modal information. Here we propose a novel dataset and benchmark suite that enables the exploration and development of large multi-modal neural network models on gene DNA sequences and natural language descriptions of gene function. We present baseline performance on benchmarks for both unsupervised and supervised tasks that demonstrate the difficulty of this modeling objective, while demonstrating the potential benefit of incorporating multi-modal data types in function prediction compared to DNA sequences alone. Our dataset is at: https://hoarfrost-lab.github.io/BioTalk/.
Invariant Risk Minimisation for Cross-Organism Inference: Substituting Mouse Data for Human Data in Human Risk Factor Discovery
Nov 14, 2021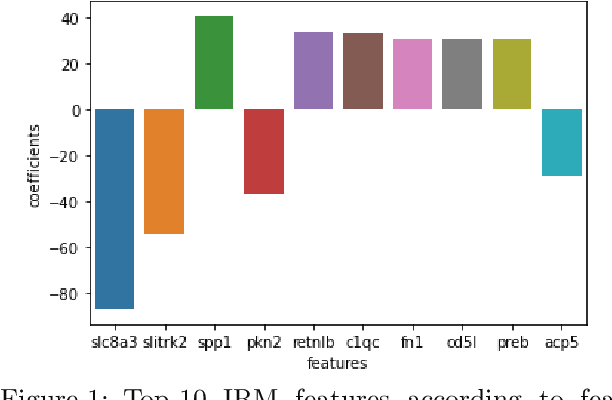
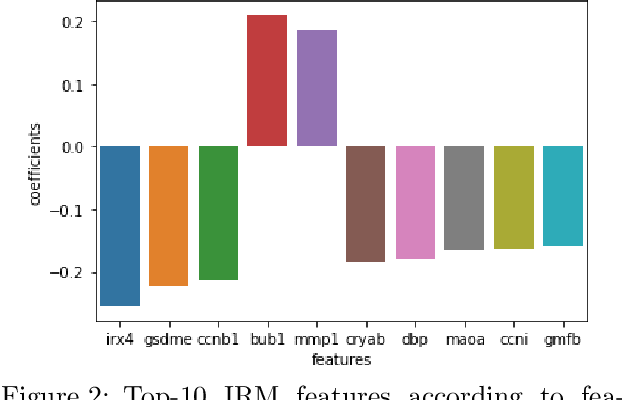
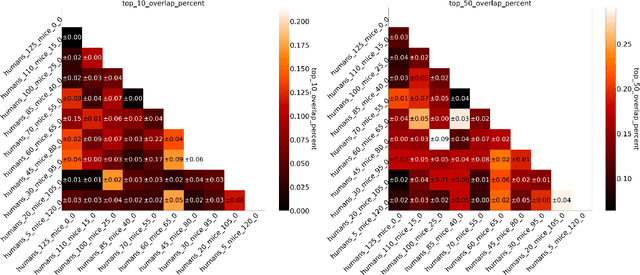
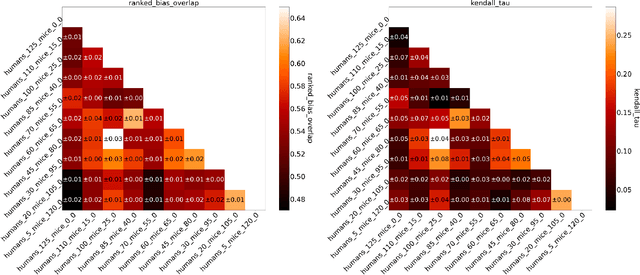
Human medical data can be challenging to obtain due to data privacy concerns, difficulties conducting certain types of experiments, or prohibitive associated costs. In many settings, data from animal models or in-vitro cell lines are available to help augment our understanding of human data. However, this data is known for having low etiological validity in comparison to human data. In this work, we augment small human medical datasets with in-vitro data and animal models. We use Invariant Risk Minimisation (IRM) to elucidate invariant features by considering cross-organism data as belonging to different data-generating environments. Our models identify genes of relevance to human cancer development. We observe a degree of consistency between varying the amounts of human and mouse data used, however, further work is required to obtain conclusive insights. As a secondary contribution, we enhance existing open source datasets and provide two uniformly processed, cross-organism, homologue gene-matched datasets to the community.