 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePatch Hierarchical Attention Transformer for Efficient Particle Jet Tagging
May 20, 2026Real-time jet tagging is critical for identifying short-lived particle decays in the high-throughput detectors of the Large Hadron Collider, where real-time trigger systems responsible for deciding which collision events to store impose strict latency and accuracy constraints. While transformer architectures achieve the highest jet tagging accuracy when compute is unconstrained, their quadratic self-attention cost makes inference restrictive on trigger budget. Existing efficient variants reduce the computational cost, but hinder the classification performance. To address this limitation, we introduce the Patch Hierarchical Attention Transformer (PHAT-JeT), which combines two mechanisms: a physics-inspired geometric message-passing module that encodes local detector-plane structure, and a hierarchical patch-based attention scheme that computes exact attention within small particle groups while preserving global context through lightweight patch-token communication. Within a restricted budget, PHAT-JeT achieves state-of-the-art accuracy and background rejection among all resource-constrained jet tagging models on four benchmarks (\textsc{hls4ml}, JetClass, Top Tagging, and Quark--Gluon). Our code is available at https://github.com/aaronw5/PHAT-JeT.
Interpreting Transformers for Jet Tagging
Dec 04, 2024Machine learning (ML) algorithms, particularly attention-based transformer models, have become indispensable for analyzing the vast data generated by particle physics experiments like ATLAS and CMS at the CERN LHC. Particle Transformer (ParT), a state-of-the-art model, leverages particle-level attention to improve jet-tagging tasks, which are critical for identifying particles resulting from proton collisions. This study focuses on interpreting ParT by analyzing attention heat maps and particle-pair correlations on the $\eta$-$\phi$ plane, revealing a binary attention pattern where each particle attends to at most one other particle. At the same time, we observe that ParT shows varying focus on important particles and subjets depending on decay, indicating that the model learns traditional jet substructure observables. These insights enhance our understanding of the model's internal workings and learning process, offering potential avenues for improving the efficiency of transformer architectures in future high-energy physics applications.
Deep Learning Methods for Protein Family Classification on PDB Sequencing Data
Jul 14, 2022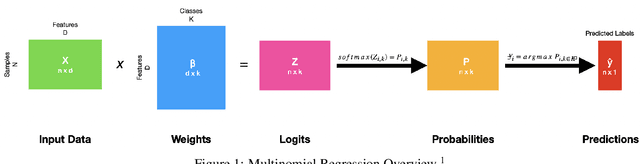
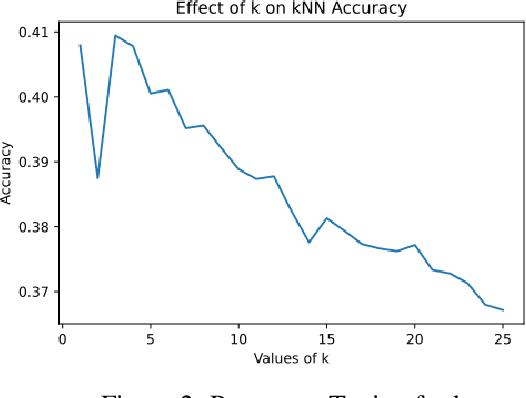
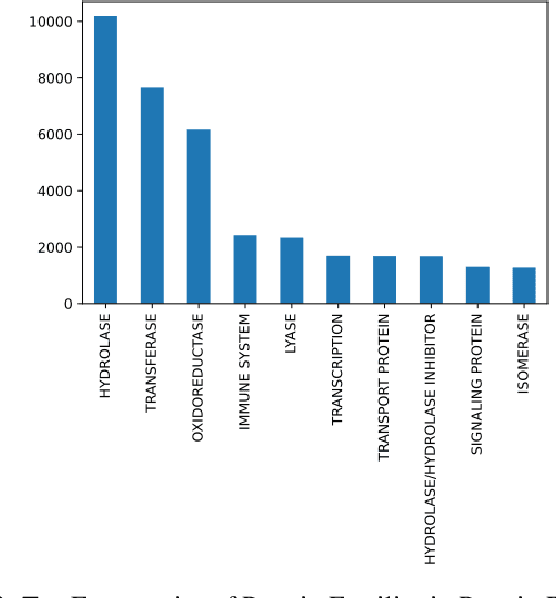
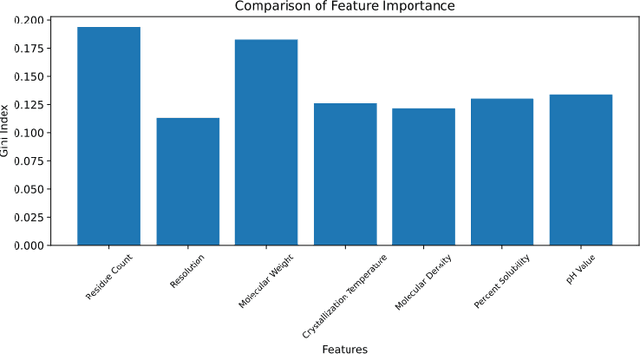
Composed of amino acid chains that influence how they fold and thus dictating their function and features, proteins are a class of macromolecules that play a central role in major biological processes and are required for the structure, function, and regulation of the body's tissues. Understanding protein functions is vital to the development of therapeutics and precision medicine, and hence the ability to classify proteins and their functions based on measurable features is crucial; indeed, the automatic inference of a protein's properties from its sequence of amino acids, known as its primary structure, remains an important open problem within the field of bioinformatics, especially given the recent advancements in sequencing technologies and the extensive number of known but uncategorized proteins with unknown properties. In this work, we demonstrate and compare the performance of several deep learning frameworks, including novel bi-directional LSTM and convolutional models, on widely available sequencing data from the Protein Data Bank (PDB) of the Research Collaboratory for Structural Bioinformatics (RCSB), as well as benchmark this performance against classical machine learning approaches, including k-nearest neighbors and multinomial regression classifiers, trained on experimental data. Our results show that our deep learning models deliver superior performance to classical machine learning methods, with the convolutional architecture providing the most impressive inference performance.
Ultra-low latency recurrent neural network inference on FPGAs for physics applications with hls4ml
Jul 01, 2022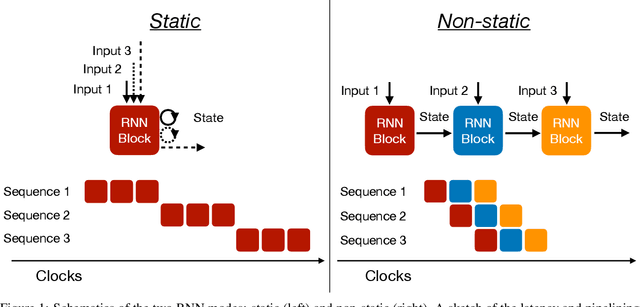
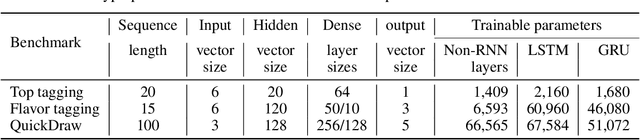
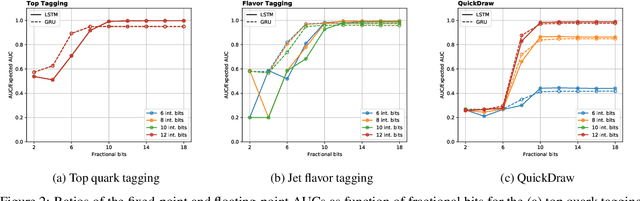

Recurrent neural networks have been shown to be effective architectures for many tasks in high energy physics, and thus have been widely adopted. Their use in low-latency environments has, however, been limited as a result of the difficulties of implementing recurrent architectures on field-programmable gate arrays (FPGAs). In this paper we present an implementation of two types of recurrent neural network layers -- long short-term memory and gated recurrent unit -- within the hls4ml framework. We demonstrate that our implementation is capable of producing effective designs for both small and large models, and can be customized to meet specific design requirements for inference latencies and FPGA resources. We show the performance and synthesized designs for multiple neural networks, many of which are trained specifically for jet identification tasks at the CERN Large Hadron Collider.
SimpleChrome: Encoding of Combinatorial Effects for Predicting Gene Expression
Dec 17, 2020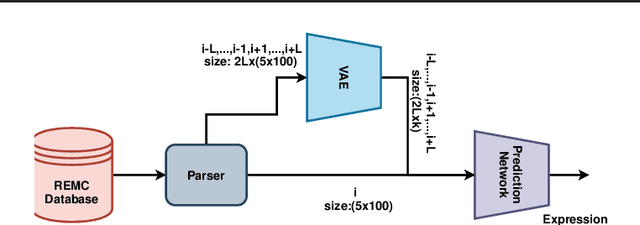
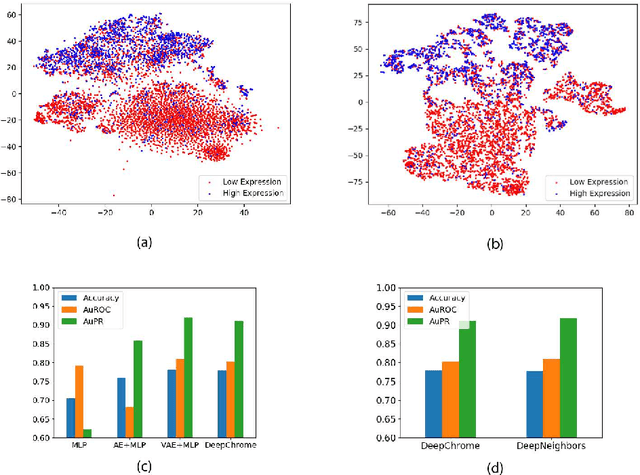
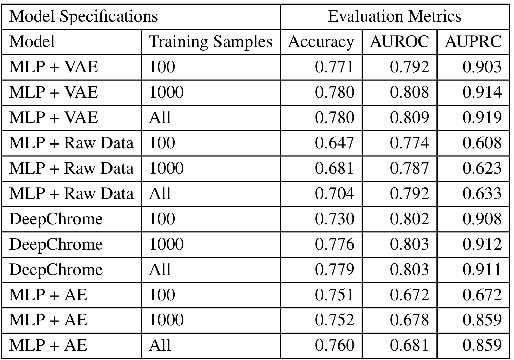
Due to recent breakthroughs in state-of-the-art DNA sequencing technology, genomics data sets have become ubiquitous. The emergence of large-scale data sets provides great opportunities for better understanding of genomics, especially gene regulation. Although each cell in the human body contains the same set of DNA information, gene expression controls the functions of these cells by either turning genes on or off, known as gene expression levels. There are two important factors that control the expression level of each gene: (1) Gene regulation such as histone modifications can directly regulate gene expression. (2) Neighboring genes that are functionally related to or interact with each other that can also affect gene expression level. Previous efforts have tried to address the former using Attention-based model. However, addressing the second problem requires the incorporation of all potentially related gene information into the model. Though modern machine learning and deep learning models have been able to capture gene expression signals when applied to moderately sized data, they have struggled to recover the underlying signals of the data due to the nature of the data's higher dimensionality. To remedy this issue, we present SimpleChrome, a deep learning model that learns the latent histone modification representations of genes. The features learned from the model allow us to better understand the combinatorial effects of cross-gene interactions and direct gene regulation on the target gene expression. The results of this paper show outstanding improvements on the predictive capabilities of downstream models and greatly relaxes the need for a large data set to learn a robust, generalized neural network. These results have immediate downstream effects in epigenomics research and drug development.