 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeK-Paths: Reasoning over Graph Paths for Drug Repurposing and Drug Interaction Prediction
Feb 18, 2025Drug discovery is a complex and time-intensive process that requires identifying and validating new therapeutic candidates. Computational approaches using large-scale biomedical knowledge graphs (KGs) offer a promising solution to accelerate this process. However, extracting meaningful insights from large-scale KGs remains challenging due to the complexity of graph traversal. Existing subgraph-based methods are tailored to graph neural networks (GNNs), making them incompatible with other models, such as large language models (LLMs). We introduce K-Paths, a retrieval framework that extracts structured, diverse, and biologically meaningful paths from KGs. Integrating these paths enables LLMs and GNNs to effectively predict unobserved drug-drug and drug-disease interactions. Unlike traditional path-ranking approaches, K-Paths retrieves and transforms paths into a structured format that LLMs can directly process, facilitating explainable reasoning. K-Paths employs a diversity-aware adaptation of Yen's algorithm to retrieve the K shortest loopless paths between entities in an interaction query, prioritizing biologically relevant and diverse relationships. Our experiments on benchmark datasets show that K-Paths improves the zero-shot performance of Llama 8.1B's F1-score by 12.45 points on drug repurposing and 13.42 points on interaction severity prediction. We also show that Llama 70B achieves F1-score gains of 6.18 and 8.46 points, respectively. K-Paths also improves the supervised training efficiency of EmerGNN, a state-of-the-art GNN, by reducing KG size by 90% while maintaining strong predictive performance. Beyond its scalability and efficiency, K-Paths uniquely bridges the gap between KGs and LLMs, providing explainable rationales for predicted interactions. These capabilities show that K-Paths is a valuable tool for efficient data-driven drug discovery.
SimpleChrome: Encoding of Combinatorial Effects for Predicting Gene Expression
Dec 17, 2020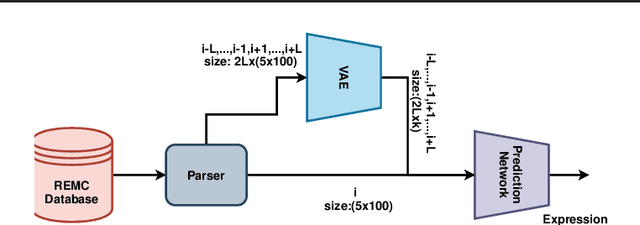
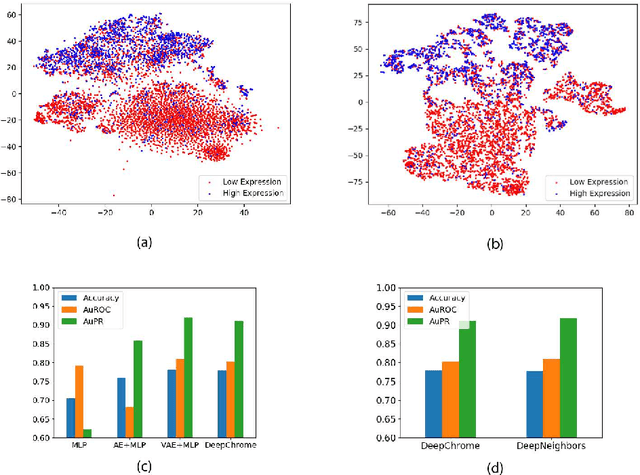
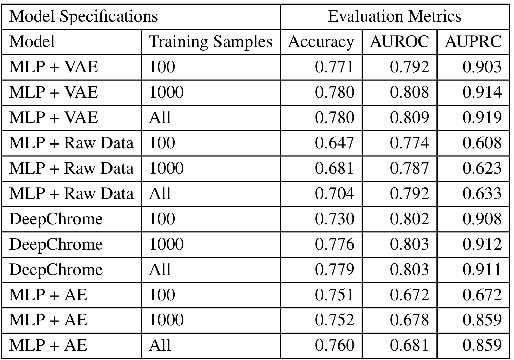
Due to recent breakthroughs in state-of-the-art DNA sequencing technology, genomics data sets have become ubiquitous. The emergence of large-scale data sets provides great opportunities for better understanding of genomics, especially gene regulation. Although each cell in the human body contains the same set of DNA information, gene expression controls the functions of these cells by either turning genes on or off, known as gene expression levels. There are two important factors that control the expression level of each gene: (1) Gene regulation such as histone modifications can directly regulate gene expression. (2) Neighboring genes that are functionally related to or interact with each other that can also affect gene expression level. Previous efforts have tried to address the former using Attention-based model. However, addressing the second problem requires the incorporation of all potentially related gene information into the model. Though modern machine learning and deep learning models have been able to capture gene expression signals when applied to moderately sized data, they have struggled to recover the underlying signals of the data due to the nature of the data's higher dimensionality. To remedy this issue, we present SimpleChrome, a deep learning model that learns the latent histone modification representations of genes. The features learned from the model allow us to better understand the combinatorial effects of cross-gene interactions and direct gene regulation on the target gene expression. The results of this paper show outstanding improvements on the predictive capabilities of downstream models and greatly relaxes the need for a large data set to learn a robust, generalized neural network. These results have immediate downstream effects in epigenomics research and drug development.