 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeDeep Learning Methods for Protein Family Classification on PDB Sequencing Data
Paper and Code
Jul 14, 2022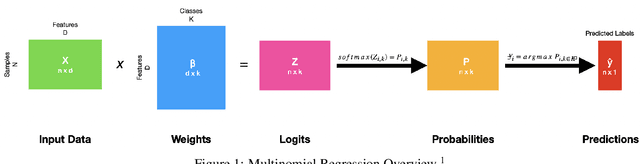
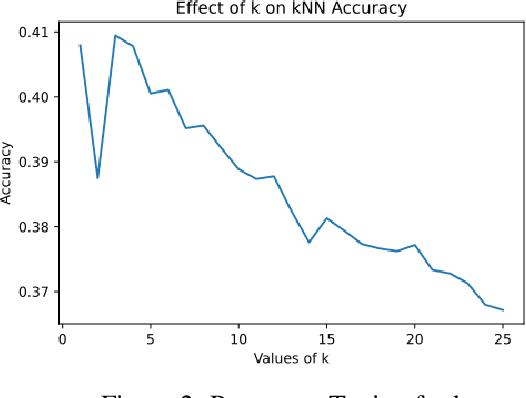
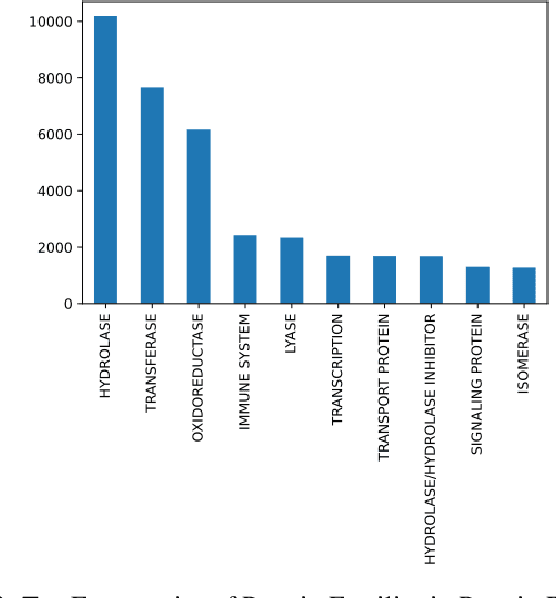
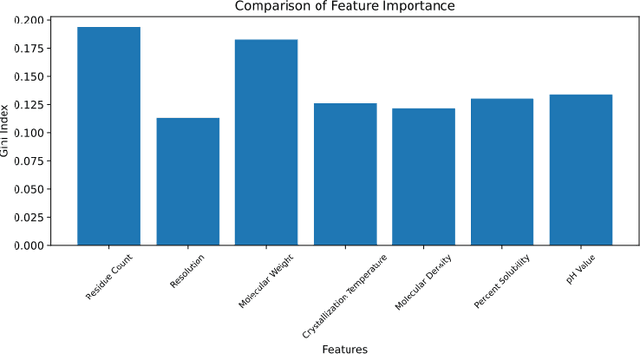
Composed of amino acid chains that influence how they fold and thus dictating their function and features, proteins are a class of macromolecules that play a central role in major biological processes and are required for the structure, function, and regulation of the body's tissues. Understanding protein functions is vital to the development of therapeutics and precision medicine, and hence the ability to classify proteins and their functions based on measurable features is crucial; indeed, the automatic inference of a protein's properties from its sequence of amino acids, known as its primary structure, remains an important open problem within the field of bioinformatics, especially given the recent advancements in sequencing technologies and the extensive number of known but uncategorized proteins with unknown properties. In this work, we demonstrate and compare the performance of several deep learning frameworks, including novel bi-directional LSTM and convolutional models, on widely available sequencing data from the Protein Data Bank (PDB) of the Research Collaboratory for Structural Bioinformatics (RCSB), as well as benchmark this performance against classical machine learning approaches, including k-nearest neighbors and multinomial regression classifiers, trained on experimental data. Our results show that our deep learning models deliver superior performance to classical machine learning methods, with the convolutional architecture providing the most impressive inference performance.