 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeStructure-Aware E(3)-Invariant Molecular Conformer Aggregation Networks
Paper and Code
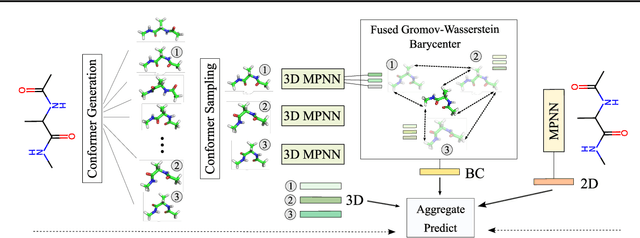

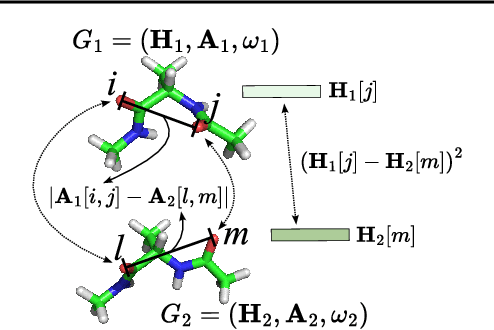

A molecule's 2D representation consists of its atoms, their attributes, and the molecule's covalent bonds. A 3D (geometric) representation of a molecule is called a conformer and consists of its atom types and Cartesian coordinates. Every conformer has a potential energy, and the lower this energy, the more likely it occurs in nature. Most existing machine learning methods for molecular property prediction consider either 2D molecular graphs or 3D conformer structure representations in isolation. Inspired by recent work on using ensembles of conformers in conjunction with 2D graph representations, we propose E(3)-invariant molecular conformer aggregation networks. The method integrates a molecule's 2D representation with that of multiple of its conformers. Contrary to prior work, we propose a novel 2D--3D aggregation mechanism based on a differentiable solver for the \emph{Fused Gromov-Wasserstein Barycenter} problem and the use of an efficient online conformer generation method based on distance geometry. We show that the proposed aggregation mechanism is E(3) invariant and provides an efficient GPU implementation. Moreover, we demonstrate that the aggregation mechanism helps to outperform state-of-the-art property prediction methods on established datasets significantly.