 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeChinese Labor Law Large Language Model Benchmark
Jan 15, 2026Recent advances in large language models (LLMs) have led to substantial progress in domain-specific applications, particularly within the legal domain. However, general-purpose models such as GPT-4 often struggle with specialized subdomains that require precise legal knowledge, complex reasoning, and contextual sensitivity. To address these limitations, we present LabourLawLLM, a legal large language model tailored to Chinese labor law. We also introduce LabourLawBench, a comprehensive benchmark covering diverse labor-law tasks, including legal provision citation, knowledge-based question answering, case classification, compensation computation, named entity recognition, and legal case analysis. Our evaluation framework combines objective metrics (e.g., ROUGE-L, accuracy, F1, and soft-F1) with subjective assessment based on GPT-4 scoring. Experiments show that LabourLawLLM consistently outperforms general-purpose and existing legal-specific LLMs across task categories. Beyond labor law, our methodology provides a scalable approach for building specialized LLMs in other legal subfields, improving accuracy, reliability, and societal value of legal AI applications.
Retrosynthesis Prediction via Search in (Hyper) Graph
Feb 09, 2024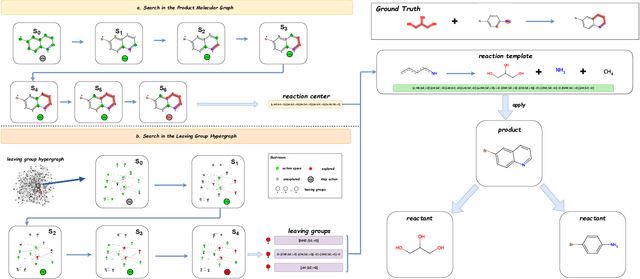
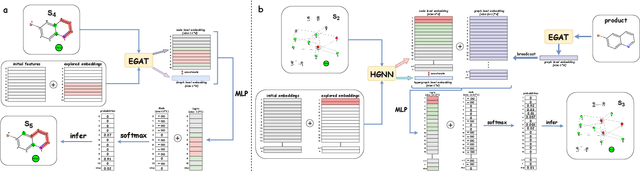
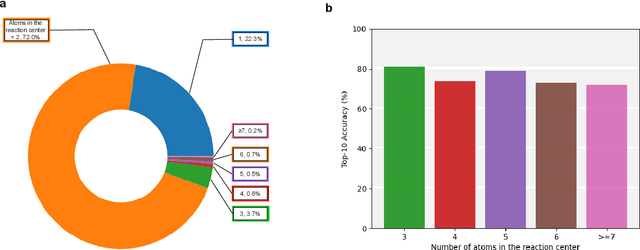
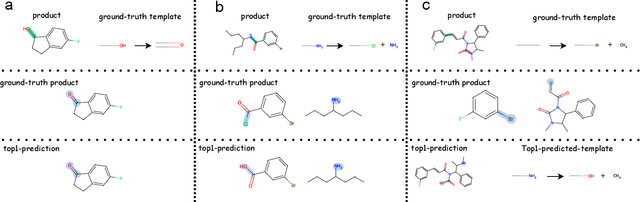
Predicting reactants from a specified core product stands as a fundamental challenge within organic synthesis, termed retrosynthesis prediction. Recently, semi-template-based methods and graph-edits-based methods have achieved good performance in terms of both interpretability and accuracy. However, due to their mechanisms these methods cannot predict complex reactions, e.g., reactions with multiple reaction center or attaching the same leaving group to more than one atom. In this study we propose a semi-template-based method, the \textbf{Retro}synthesis via \textbf{S}earch \textbf{i}n (Hyper) \textbf{G}raph (RetroSiG) framework to alleviate these limitations. In the proposed method, we turn the reaction center identification and the leaving group completion tasks as tasks of searching in the product molecular graph and leaving group hypergraph respectively. As a semi-template-based method RetroSiG has several advantages. First, RetroSiG is able to handle the complex reactions mentioned above by its novel search mechanism. Second, RetroSiG naturally exploits the hypergraph to model the implicit dependencies between leaving groups. Third, RetroSiG makes full use of the prior, i.e., one-hop constraint. It reduces the search space and enhances overall performance. Comprehensive experiments demonstrated that RetroSiG achieved competitive results. Furthermore, we conducted experiments to show the capability of RetroSiG in predicting complex reactions. Ablation experiments verified the efficacy of specific elements, such as the one-hop constraint and the leaving group hypergraph.
Use neural networks to recognize students' handwritten letters and incorrect symbols
Sep 12, 2023Correcting students' multiple-choice answers is a repetitive and mechanical task that can be considered an image multi-classification task. Assuming possible options are 'abcd' and the correct option is one of the four, some students may write incorrect symbols or options that do not exist. In this paper, five classifications were set up - four for possible correct options and one for other incorrect writing. This approach takes into account the possibility of non-standard writing options.
RCsearcher: Reaction Center Identification in Retrosynthesis via Deep Q-Learning
Jan 28, 2023The reaction center consists of atoms in the product whose local properties are not identical to the corresponding atoms in the reactants. Prior studies on reaction center identification are mainly on semi-templated retrosynthesis methods. Moreover, they are limited to single reaction center identification. However, many reaction centers are comprised of multiple bonds or atoms in reality. We refer to it as the multiple reaction center. This paper presents RCsearcher, a unified framework for single and multiple reaction center identification that combines the advantages of the graph neural network and deep reinforcement learning. The critical insight in this framework is that the single or multiple reaction center must be a node-induced subgraph of the molecular product graph. At each step, it considers choosing one node in the molecular product graph and adding it to the explored node-induced subgraph as an action. Comprehensive experiments demonstrate that RCsearcher consistently outperforms other baselines and can extrapolate the reaction center patterns that have not appeared in the training set. Ablation experiments verify the effectiveness of individual components, including the beam search and one-hop constraint of action space.
More Interpretable Graph Similarity Computation via Maximum Common Subgraph Inference
Aug 25, 2022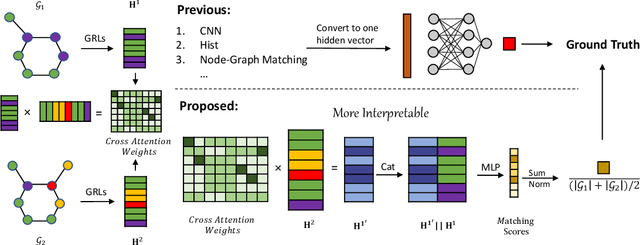



Graph similarity measurement, which computes the distance/similarity between two graphs, arises in various graph-related tasks. Recent learning-based methods lack interpretability, as they directly transform interaction information between two graphs into one hidden vector and then map it to similarity. To cope with this problem, this study proposes a more interpretable end-to-end paradigm for graph similarity learning, named Similarity Computation via Maximum Common Subgraph Inference (INFMCS). Our critical insight into INFMCS is the strong correlation between similarity score and Maximum Common Subgraph (MCS). We implicitly infer MCS to obtain the normalized MCS size, with the supervision information being only the similarity score during training. To capture more global information, we also stack some vanilla transformer encoder layers with graph convolution layers and propose a novel permutation-invariant node Positional Encoding. The entire model is quite simple yet effective. Comprehensive experiments demonstrate that INFMCS consistently outperforms state-of-the-art baselines for graph-graph classification and regression tasks. Ablation experiments verify the effectiveness of the proposed computation paradigm and other components. Also, visualization and statistics of results reveal the interpretability of INFMCS.
Sub-GMN: The Subgraph Matching Network Model
Apr 30, 2021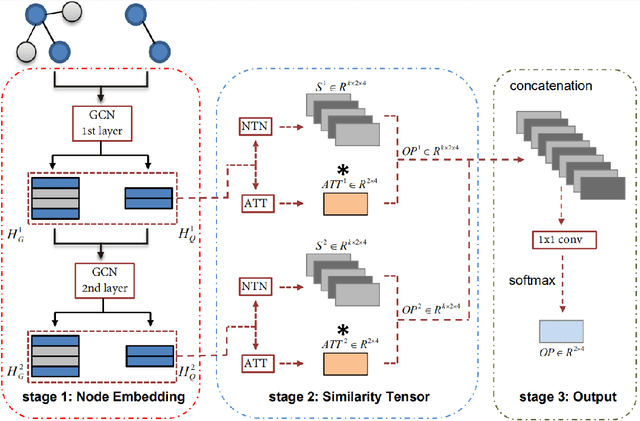


As one of the most fundamental tasks in graph theory, subgraph matching is a crucial task in many fields, ranging from information retrieval, computer vision, biology, chemistry and natural language processing. Yet subgraph matching problem remains to be an NP-complete problem. This study proposes an end-to-end learning-based approximate method for subgraph matching task, called subgraph matching network (Sub-GMN). The proposed Sub-GMN firstly uses graph representation learning to map nodes to node-level embedding. It then combines metric learning and attention mechanisms to model the relationship between matched nodes in the data graph and query graph. To test the performance of the proposed method, we applied our method on two databases. We used two existing methods, GNN and FGNN as baseline for comparison. Our experiment shows that, on dataset 1, on average the accuracy of Sub-GMN are 12.21\% and 3.2\% higher than that of GNN and FGNN respectively. On average running time Sub-GMN runs 20-40 times faster than FGNN. In addition, the average F1-score of Sub-GMN on all experiments with dataset 2 reached 0.95, which demonstrates that Sub-GMN outputs more correct node-to-node matches. Comparing with the previous GNNs-based methods for subgraph matching task, our proposed Sub-GMN allows varying query and data graphes in the test/application stage, while most previous GNNs-based methods can only find a matched subgraph in the data graph during the test/application for the same query graph used in the training stage. Another advantage of our proposed Sub-GMN is that it can output a list of node-to-node matches, while most existing end-to-end GNNs based methods cannot provide the matched node pairs.