 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeRetrosynthesis Prediction via Search in (Hyper) Graph
Feb 09, 2024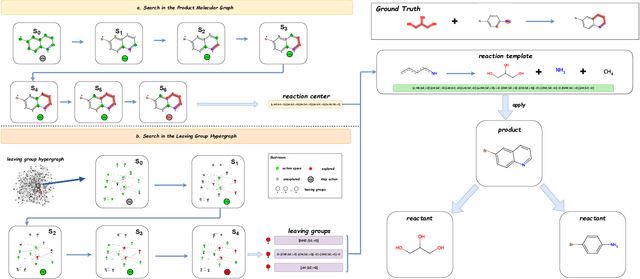
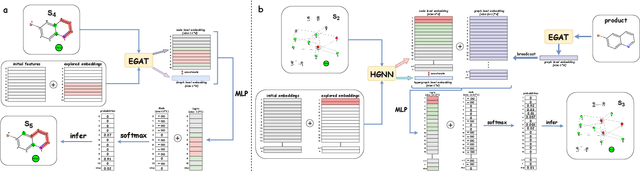
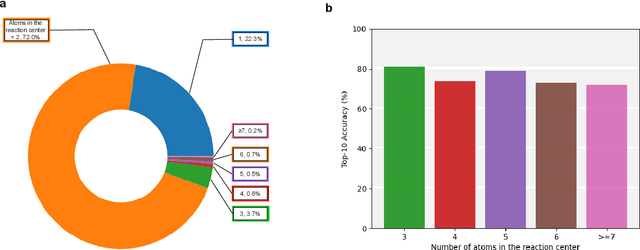
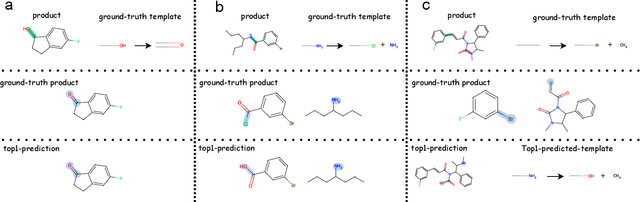
Predicting reactants from a specified core product stands as a fundamental challenge within organic synthesis, termed retrosynthesis prediction. Recently, semi-template-based methods and graph-edits-based methods have achieved good performance in terms of both interpretability and accuracy. However, due to their mechanisms these methods cannot predict complex reactions, e.g., reactions with multiple reaction center or attaching the same leaving group to more than one atom. In this study we propose a semi-template-based method, the \textbf{Retro}synthesis via \textbf{S}earch \textbf{i}n (Hyper) \textbf{G}raph (RetroSiG) framework to alleviate these limitations. In the proposed method, we turn the reaction center identification and the leaving group completion tasks as tasks of searching in the product molecular graph and leaving group hypergraph respectively. As a semi-template-based method RetroSiG has several advantages. First, RetroSiG is able to handle the complex reactions mentioned above by its novel search mechanism. Second, RetroSiG naturally exploits the hypergraph to model the implicit dependencies between leaving groups. Third, RetroSiG makes full use of the prior, i.e., one-hop constraint. It reduces the search space and enhances overall performance. Comprehensive experiments demonstrated that RetroSiG achieved competitive results. Furthermore, we conducted experiments to show the capability of RetroSiG in predicting complex reactions. Ablation experiments verified the efficacy of specific elements, such as the one-hop constraint and the leaving group hypergraph.
RCsearcher: Reaction Center Identification in Retrosynthesis via Deep Q-Learning
Jan 28, 2023The reaction center consists of atoms in the product whose local properties are not identical to the corresponding atoms in the reactants. Prior studies on reaction center identification are mainly on semi-templated retrosynthesis methods. Moreover, they are limited to single reaction center identification. However, many reaction centers are comprised of multiple bonds or atoms in reality. We refer to it as the multiple reaction center. This paper presents RCsearcher, a unified framework for single and multiple reaction center identification that combines the advantages of the graph neural network and deep reinforcement learning. The critical insight in this framework is that the single or multiple reaction center must be a node-induced subgraph of the molecular product graph. At each step, it considers choosing one node in the molecular product graph and adding it to the explored node-induced subgraph as an action. Comprehensive experiments demonstrate that RCsearcher consistently outperforms other baselines and can extrapolate the reaction center patterns that have not appeared in the training set. Ablation experiments verify the effectiveness of individual components, including the beam search and one-hop constraint of action space.