 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeProbing RLVR training instability through the lens of objective-level hacking
Feb 01, 2026Prolonged reinforcement learning with verifiable rewards (RLVR) has been shown to drive continuous improvements in the reasoning capabilities of large language models, but the training is often prone to instabilities, especially in Mixture-of-Experts (MoE) architectures. Training instability severely undermines model capability improvement, yet its underlying causes and mechanisms remain poorly understood. In this work, we introduce a principled framework for understanding RLVR instability through the lens of objective-level hacking. Unlike reward hacking, which arises from exploitable verifiers, objective-level hacking emerges from token-level credit misalignment and is manifested as system-level spurious signals in the optimization objective. Grounded in our framework, together with extensive experiments on a 30B MoE model, we trace the origin and formalize the mechanism behind a key pathological training dynamic in MoE models: the abnormal growth of the training-inference discrepancy, a phenomenon widely associated with instability but previously lacking a mechanistic explanation. These findings provide a concrete and causal account of the training dynamics underlying instabilities in MoE models, offering guidance for the design of stable RLVR algorithms.
Atomic and Subgraph-aware Bilateral Aggregation for Molecular Representation Learning
May 22, 2023
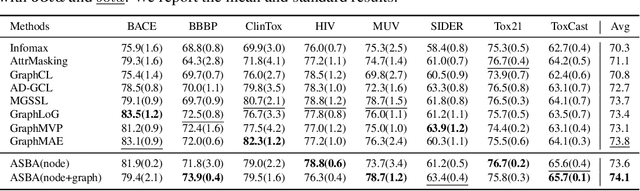
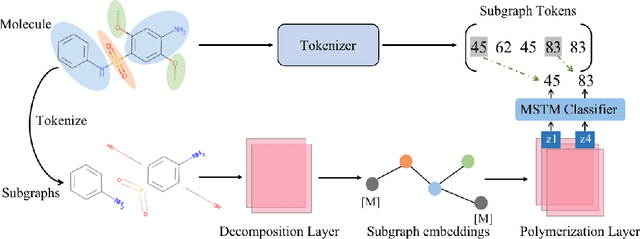
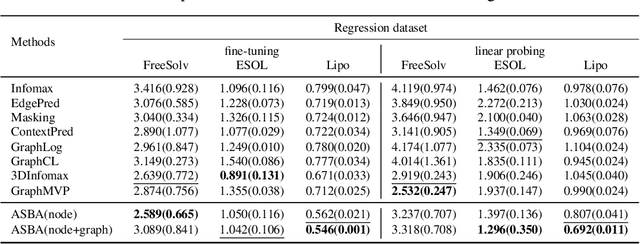
Molecular representation learning is a crucial task in predicting molecular properties. Molecules are often modeled as graphs where atoms and chemical bonds are represented as nodes and edges, respectively, and Graph Neural Networks (GNNs) have been commonly utilized to predict atom-related properties, such as reactivity and solubility. However, functional groups (subgraphs) are closely related to some chemical properties of molecules, such as efficacy, and metabolic properties, which cannot be solely determined by individual atoms. In this paper, we introduce a new model for molecular representation learning called the Atomic and Subgraph-aware Bilateral Aggregation (ASBA), which addresses the limitations of previous atom-wise and subgraph-wise models by incorporating both types of information. ASBA consists of two branches, one for atom-wise information and the other for subgraph-wise information. Considering existing atom-wise GNNs cannot properly extract invariant subgraph features, we propose a decomposition-polymerization GNN architecture for the subgraph-wise branch. Furthermore, we propose cooperative node-level and graph-level self-supervised learning strategies for ASBA to improve its generalization. Our method offers a more comprehensive way to learn representations for molecular property prediction and has broad potential in drug and material discovery applications. Extensive experiments have demonstrated the effectiveness of our method.