 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeBridging the gap between target-based and cell-based drug discovery with a graph generative multi-task model
Aug 09, 2022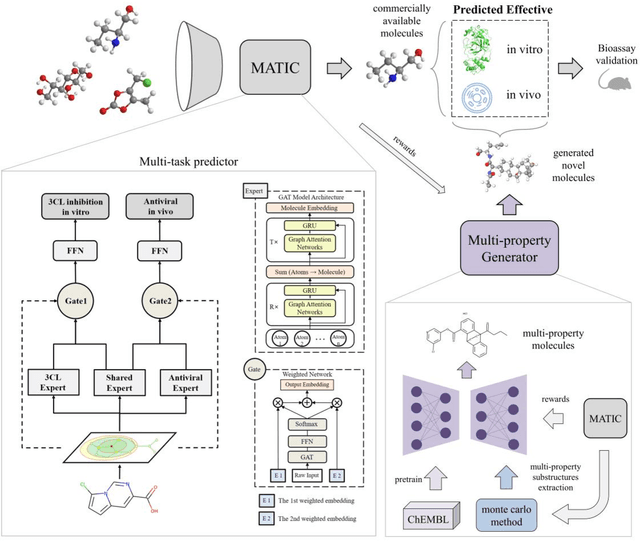
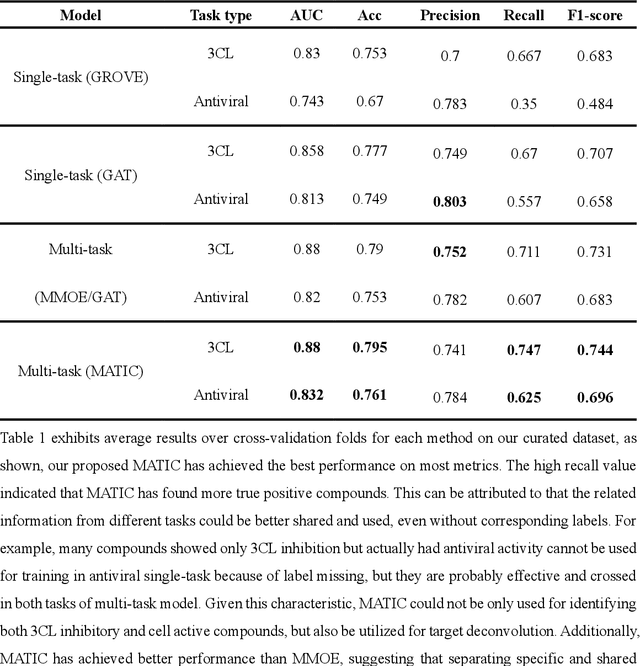
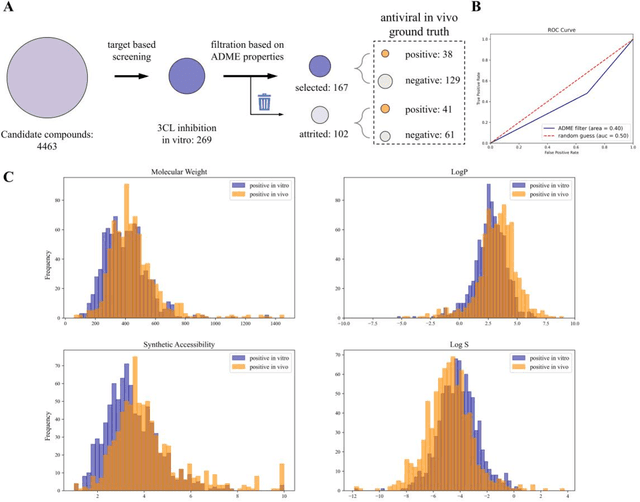
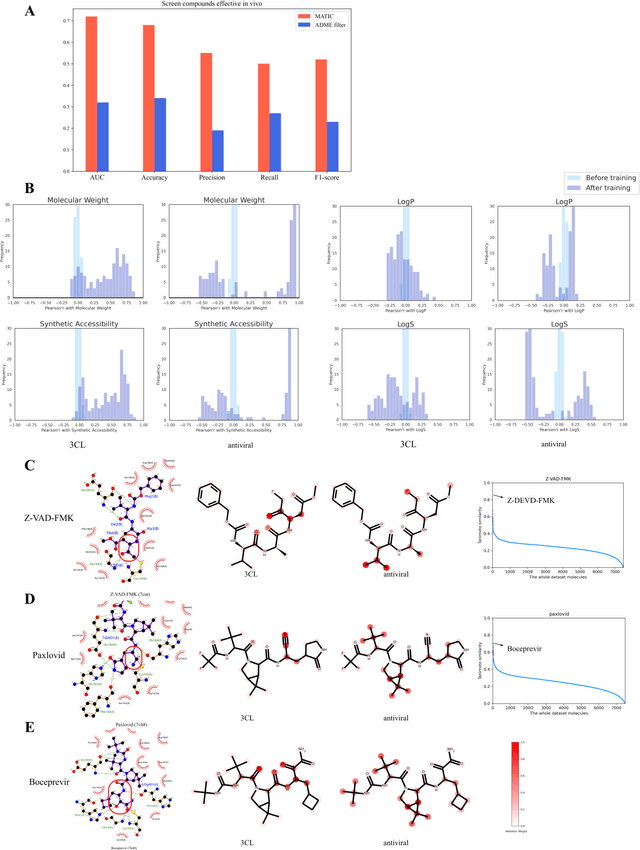
Drug discovery is vitally important for protecting human against disease. Target-based screening is one of the most popular methods to develop new drugs in the past several decades. This method efficiently screens candidate drugs inhibiting target protein in vitro, but it often fails due to inadequate activity of the selected drugs in vivo. Accurate computational methods are needed to bridge this gap. Here, we propose a novel graph multi task deep learning model to identify compounds carrying both target inhibitory and cell active (MATIC) properties. On a carefully curated SARS-CoV-2 dataset, the proposed MATIC model shows advantages comparing with traditional method in screening effective compounds in vivo. Next, we explored the model interpretability and found that the learned features for target inhibition (in vitro) or cell active (in vivo) tasks are different with molecular property correlations and atom functional attentions. Based on these findings, we utilized a monte carlo based reinforcement learning generative model to generate novel multi-property compounds with both in vitro and in vivo efficacy, thus bridging the gap between target-based and cell-based drug discovery.
A Novel Framework Integrating AI Model and Enzymological Experiments Promotes Identification of SARS-CoV-2 3CL Protease Inhibitors and Activity-based Probe
May 29, 2021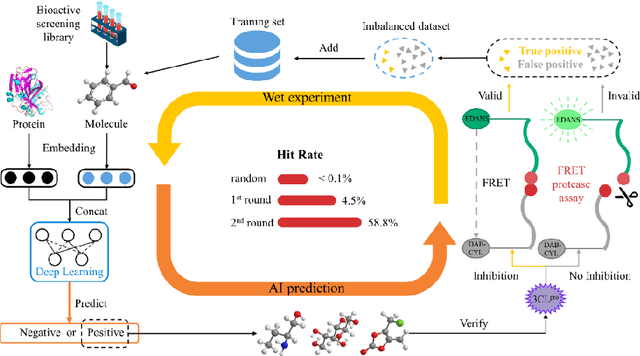
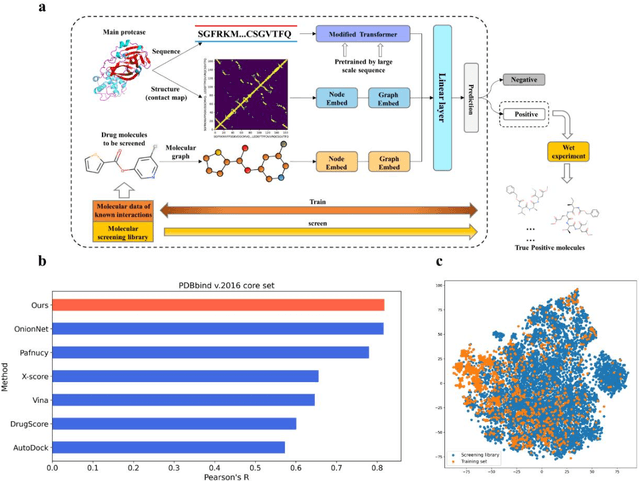
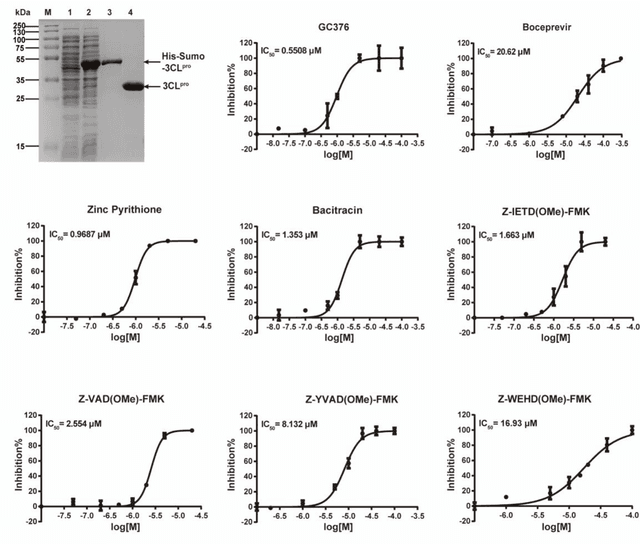
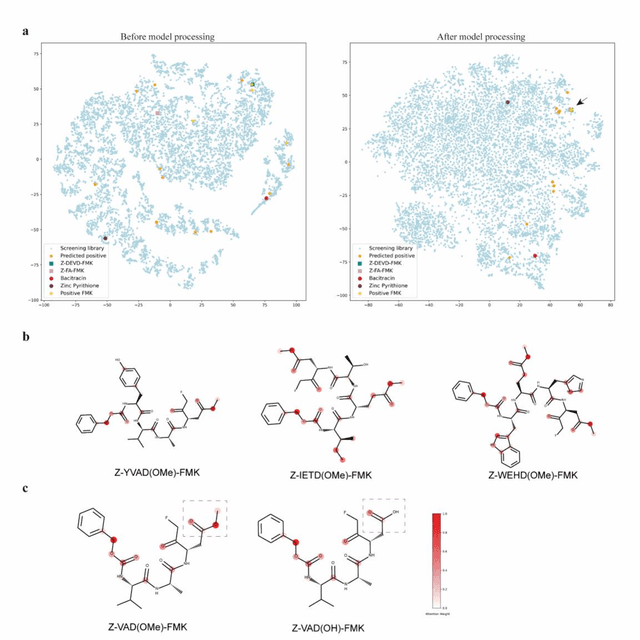
The identification of protein-ligand interaction plays a key role in biochemical research and drug discovery. Although deep learning has recently shown great promise in discovering new drugs, there remains a gap between deep learning-based and experimental approaches. Here we propose a novel framework, named AIMEE, integrating AI Model and Enzymology Experiments, to identify inhibitors against 3CL protease of SARS-CoV-2, which has taken a significant toll on people across the globe. From a bioactive chemical library, we have conducted two rounds of experiments and identified six novel inhibitors with a hit rate of 29.41%, and four of them showed an IC50 value less than 3 {\mu}M. Moreover, we explored the interpretability of the central model in AIMEE, mapping the deep learning extracted features to domain knowledge of chemical properties. Based on this knowledge, a commercially available compound was selected and proven to be an activity-based probe of 3CLpro. This work highlights the great potential of combining deep learning models and biochemical experiments for intelligent iteration and expanding the boundaries of drug discovery.