 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeSupporting Workflow Reproducibility by Linking Bioinformatics Tools across Papers and Executable Code
Mar 09, 2026Motivation: The rapid growth of biological data has intensified the need for transparent, reproducible, and well-documented computational workflows. The ability to clearly connect the steps of a workflow in the code with their description in a paper would improve workflow understanding, support reproducibility, and facilitate reuse. This task requires the linking of Bioinformatics tools in workflow code with their mentions in a published workflow description. Results: We present CoPaLink, an automated approach that integrates three components: Named Entity Recognition (NER) for identifying tool mentions in scientific text, NER for tool mentions in workflow code, and entity linking grounded on Bioinformatics knowledge bases. We propose approaches for all three steps achieving a high individual F1-measure (84 - 89) and a joint accuracy of 66 when evaluated on Nextflow workflows using Bioconda and Bioweb Knowledge bases. CoPaLink leverages corpora of scientific articles and workflow executable code with curated tool annotations to bridge the gap between narrative descriptions and workflow implementations. Availability: The code is available at https://gitlab.liris.cnrs.fr/sharefair/copalink-experiments and https://gitlab.liris.cnrs.fr/sharefair/copalink. The corpora are also available at https://doi.org/10.5281/zenodo.18526700, https://doi.org/10.5281/zenodo.18526760 and https://doi.org/10.5281/zenodo.18543814.
Knowledge-augmented Pre-trained Language Models for Biomedical Relation Extraction
May 01, 2025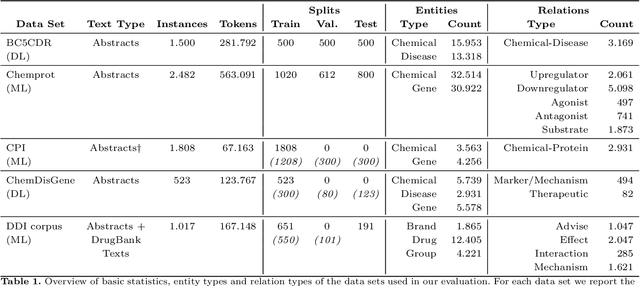
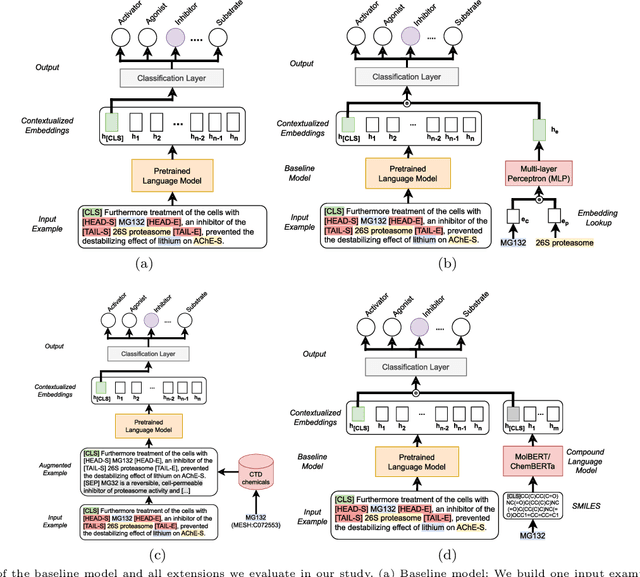
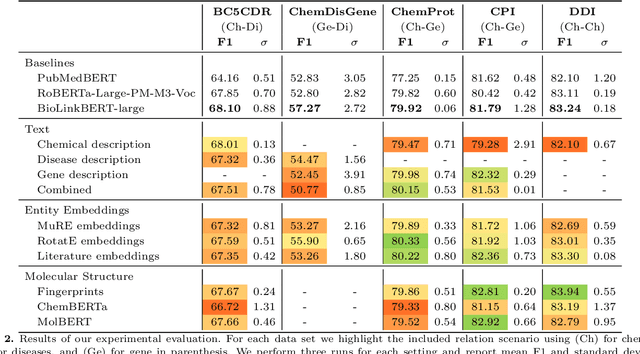
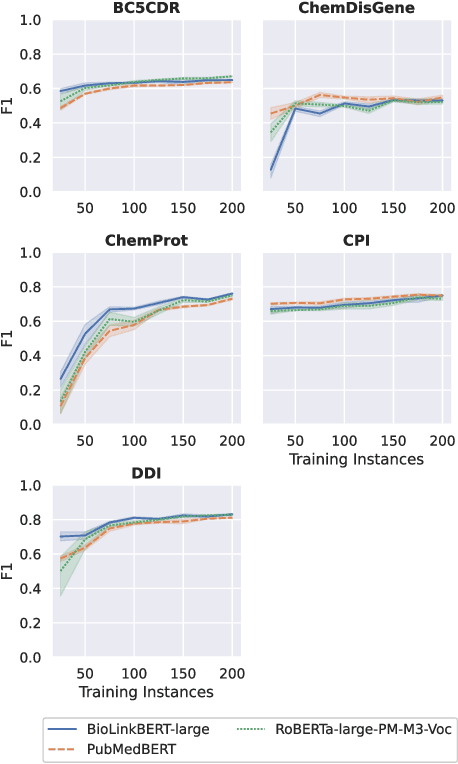
Automatic relationship extraction (RE) from biomedical literature is critical for managing the vast amount of scientific knowledge produced each year. In recent years, utilizing pre-trained language models (PLMs) has become the prevalent approach in RE. Several studies report improved performance when incorporating additional context information while fine-tuning PLMs for RE. However, variations in the PLMs applied, the databases used for augmentation, hyper-parameter optimization, and evaluation methods complicate direct comparisons between studies and raise questions about the generalizability of these findings. Our study addresses this research gap by evaluating PLMs enhanced with contextual information on five datasets spanning four relation scenarios within a consistent evaluation framework. We evaluate three baseline PLMs and first conduct extensive hyperparameter optimization. After selecting the top-performing model, we enhance it with additional data, including textual entity descriptions, relational information from knowledge graphs, and molecular structure encodings. Our findings illustrate the importance of i) the choice of the underlying language model and ii) a comprehensive hyperparameter optimization for achieving strong extraction performance. Although inclusion of context information yield only minor overall improvements, an ablation study reveals substantial benefits for smaller PLMs when such external data was included during fine-tuning.
Enhancing Document Retrieval for Curating N-ary Relations in Knowledge Bases
Apr 14, 2025Curation of biomedical knowledge bases (KBs) relies on extracting accurate multi-entity relational facts from the literature - a process that remains largely manual and expert-driven. An essential step in this workflow is retrieving documents that can support or complete partially observed n-ary relations. We present a neural retrieval model designed to assist KB curation by identifying documents that help fill in missing relation arguments and provide relevant contextual evidence. To reduce dependence on scarce gold-standard training data, we exploit existing KB records to construct weakly supervised training sets. Our approach introduces two key technical contributions: (i) a layered contrastive loss that enables learning from noisy and incomplete relational structures, and (ii) a balanced sampling strategy that generates high-quality negatives from diverse KB records. On two biomedical retrieval benchmarks, our approach achieves state-of-the-art performance, outperforming strong baselines in NDCG@10 by 5.7 and 3.7 percentage points, respectively.
CLaP -- State Detection from Time Series
Apr 02, 2025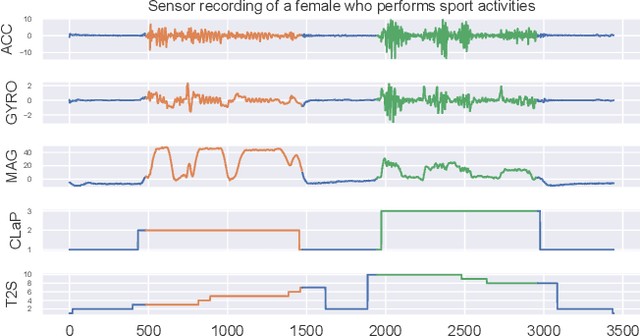
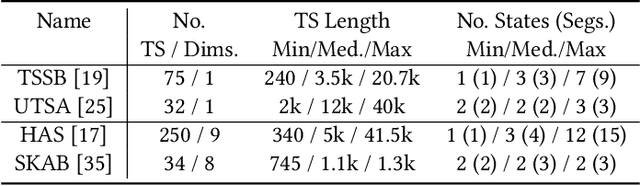
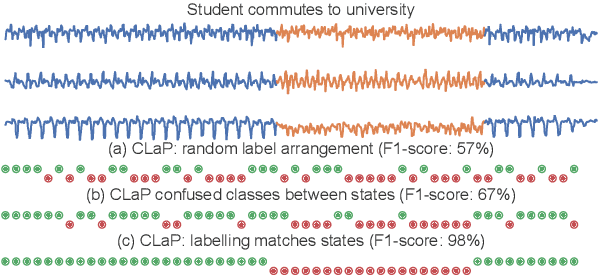
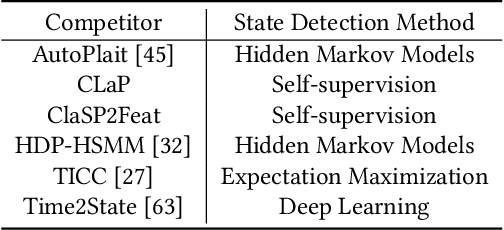
The ever-growing amount of sensor data from machines, smart devices, and the environment leads to an abundance of high-resolution, unannotated time series (TS). These recordings encode the recognizable properties of latent states and transitions from physical phenomena that can be modelled as abstract processes. The unsupervised localization and identification of these states and their transitions is the task of time series state detection (TSSD). We introduce CLaP, a new, highly accurate and efficient algorithm for TSSD. It leverages the predictive power of time series classification for TSSD in an unsupervised setting by applying novel self-supervision techniques to detect whether data segments emerge from the same state or not. To this end, CLaP cross-validates a classifier with segment-labelled subsequences to quantify confusion between segments. It merges labels from segments with high confusion, representing the same latent state, if this leads to an increase in overall classification quality. We conducted an experimental evaluation using 391 TS from four benchmarks and found CLaP to be significantly more precise in detecting states than five state-of-the-art competitors. It achieves the best accuracy-runtime tradeoff and is scalable to large TS. We provide a Python implementation of CLaP, which can be deployed in TS analysis workflows.
Discovering Leitmotifs in Multidimensional Time Series
Oct 16, 2024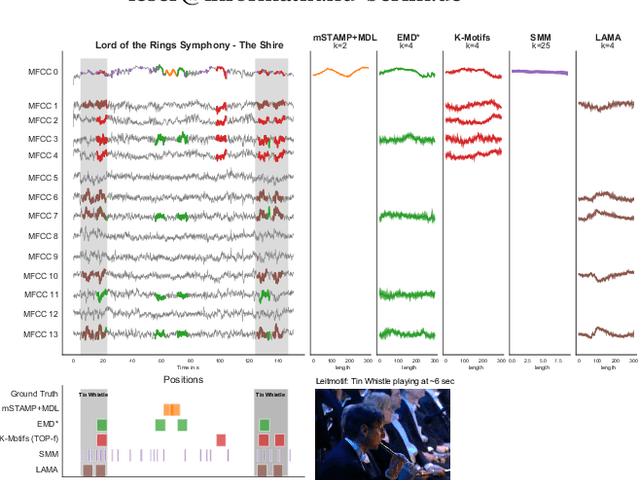
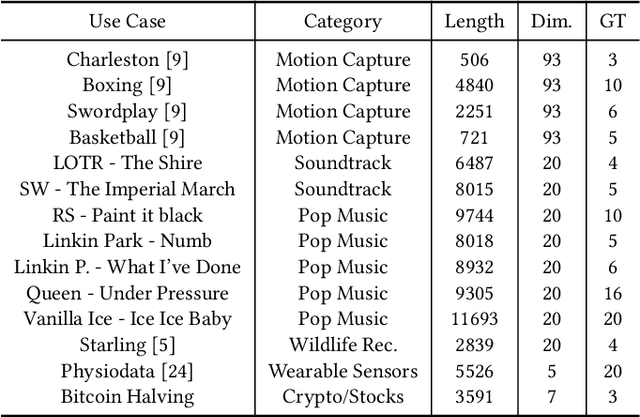
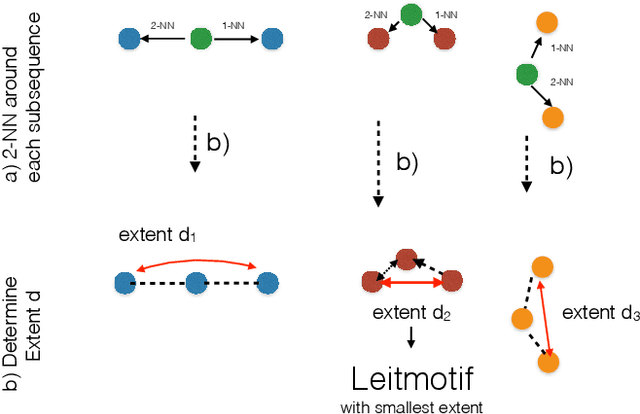
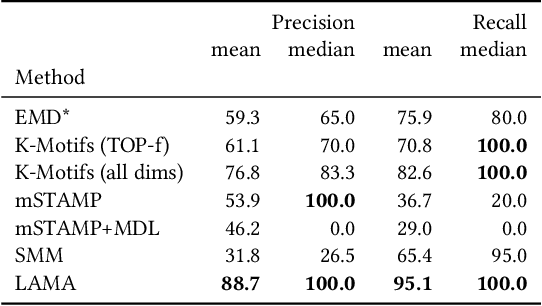
A leitmotif is a recurring theme in literature, movies or music that carries symbolic significance for the piece it is contained in. When this piece can be represented as a multi-dimensional time series (MDTS), such as acoustic or visual observations, finding a leitmotif is equivalent to the pattern discovery problem, which is an unsupervised and complex problem in time series analytics. Compared to the univariate case, it carries additional complexity because patterns typically do not occur in all dimensions but only in a few - which are, however, unknown and must be detected by the method itself. In this paper, we present the novel, efficient and highly effective leitmotif discovery algorithm LAMA for MDTS. LAMA rests on two core principals: (a) a leitmotif manifests solely given a yet unknown number of sub-dimensions - neither too few, nor too many, and (b) the set of sub-dimensions are not independent from the best pattern found therein, necessitating both problems to be approached in a joint manner. In contrast to most previous methods, LAMA tackles both problems jointly - instead of independently selecting dimensions (or leitmotifs) and finding the best leitmotifs (or dimensions). Our experimental evaluation on a novel ground-truth annotated benchmark of 14 distinct real-life data sets shows that LAMA, when compared to four state-of-the-art baselines, shows superior performance in detecting meaningful patterns without increased computational complexity.
HunFlair2 in a cross-corpus evaluation of biomedical named entity recognition and normalization tools
Feb 20, 2024

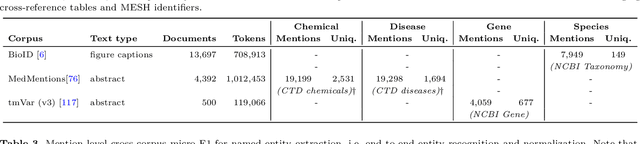

With the exponential growth of the life science literature, biomedical text mining (BTM) has become an essential technology for accelerating the extraction of insights from publications. Identifying named entities (e.g., diseases, drugs, or genes) in texts and their linkage to reference knowledge bases are crucial steps in BTM pipelines to enable information aggregation from different documents. However, tools for these two steps are rarely applied in the same context in which they were developed. Instead, they are applied in the wild, i.e., on application-dependent text collections different from those used for the tools' training, varying, e.g., in focus, genre, style, and text type. This raises the question of whether the reported performance of BTM tools can be trusted for downstream applications. Here, we report on the results of a carefully designed cross-corpus benchmark for named entity extraction, where tools were applied systematically to corpora not used during their training. Based on a survey of 28 published systems, we selected five for an in-depth analysis on three publicly available corpora encompassing four different entity types. Comparison between tools results in a mixed picture and shows that, in a cross-corpus setting, the performance is significantly lower than the one reported in an in-corpus setting. HunFlair2 showed the best performance on average, being closely followed by PubTator. Our results indicate that users of BTM tools should expect diminishing performances when applying them in the wild compared to original publications and show that further research is necessary to make BTM tools more robust.
BELHD: Improving Biomedical Entity Linking with Homonoym Disambiguation
Jan 10, 2024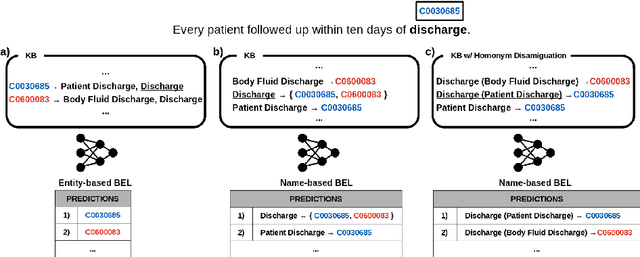
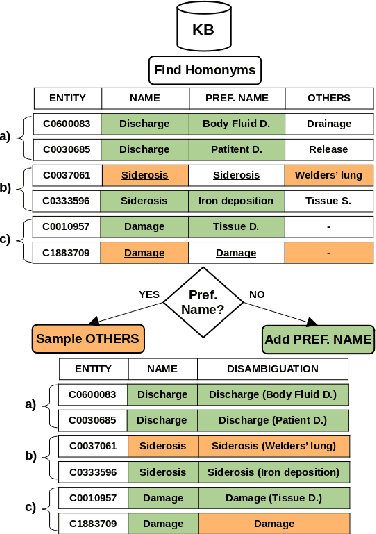
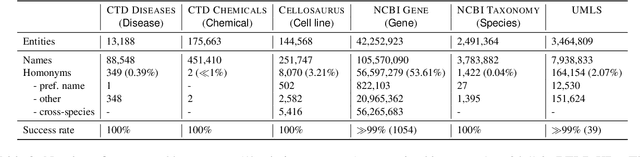
Biomedical entity linking (BEL) is the task of grounding entity mentions to a knowledge base (KB). A popular approach to the task are name-based methods, i.e. those identifying the most appropriate name in the KB for a given mention, either via dense retrieval or autoregressive modeling. However, as these methods directly return KB names, they cannot cope with homonyms, i.e. different KB entities sharing the exact same name. This significantly affects their performance, especially for KBs where homonyms account for a large amount of entity mentions (e.g. UMLS and NCBI Gene). We therefore present BELHD (Biomedical Entity Linking with Homonym Disambiguation), a new name-based method that copes with this challenge. Specifically, BELHD builds upon the BioSyn (Sung et al.,2020) model introducing two crucial extensions. First, it performs a preprocessing of the KB in which it expands homonyms with an automatically chosen disambiguating string, thus enforcing unique linking decisions. Second, we introduce candidate sharing, a novel strategy to select candidates for contrastive learning that enhances the overall training signal. Experiments with 10 corpora and five entity types show that BELHD improves upon state-of-the-art approaches, achieving the best results in 6 out 10 corpora with an average improvement of 4.55pp recall@1. Furthermore, the KB preprocessing is orthogonal to the core prediction model and thus can also improve other methods, which we exemplify for GenBioEL (Yuan et al, 2022), a generative name-based BEL approach. Code is available at: link added upon publication.
Large Language Models to the Rescue: Reducing the Complexity in Scientific Workflow Development Using ChatGPT
Nov 06, 2023
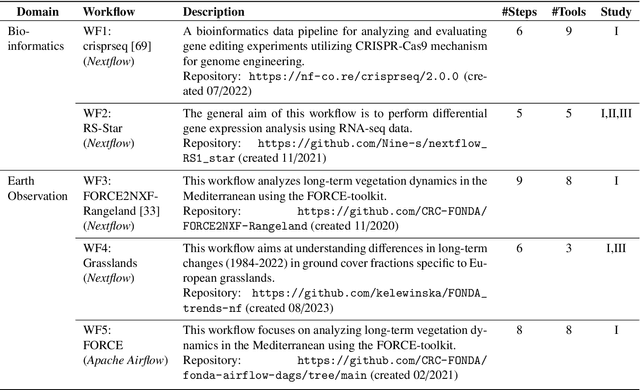

Scientific workflow systems are increasingly popular for expressing and executing complex data analysis pipelines over large datasets, as they offer reproducibility, dependability, and scalability of analyses by automatic parallelization on large compute clusters. However, implementing workflows is difficult due to the involvement of many black-box tools and the deep infrastructure stack necessary for their execution. Simultaneously, user-supporting tools are rare, and the number of available examples is much lower than in classical programming languages. To address these challenges, we investigate the efficiency of Large Language Models (LLMs), specifically ChatGPT, to support users when dealing with scientific workflows. We performed three user studies in two scientific domains to evaluate ChatGPT for comprehending, adapting, and extending workflows. Our results indicate that LLMs efficiently interpret workflows but achieve lower performance for exchanging components or purposeful workflow extensions. We characterize their limitations in these challenging scenarios and suggest future research directions.
Raising the ClaSS of Streaming Time Series Segmentation
Oct 31, 2023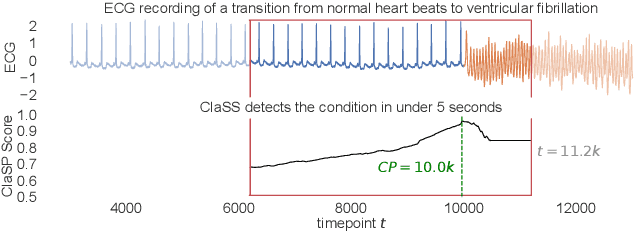


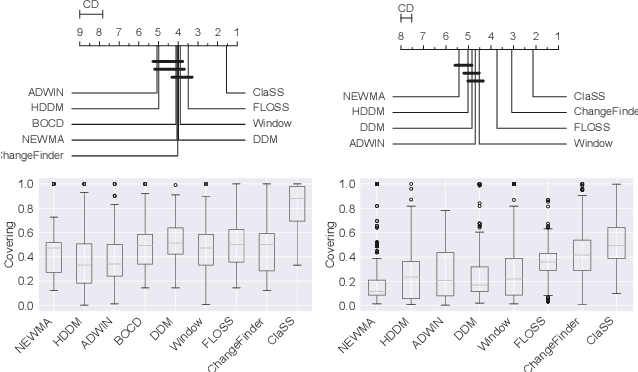
Ubiquitous sensors today emit high frequency streams of numerical measurements that reflect properties of human, animal, industrial, commercial, and natural processes. Shifts in such processes, e.g. caused by external events or internal state changes, manifest as changes in the recorded signals. The task of streaming time series segmentation (STSS) is to partition the stream into consecutive variable-sized segments that correspond to states of the observed processes or entities. The partition operation itself must in performance be able to cope with the input frequency of the signals. We introduce ClaSS, a novel, efficient, and highly accurate algorithm for STSS. ClaSS assesses the homogeneity of potential partitions using self-supervised time series classification and applies statistical tests to detect significant change points (CPs). In our experimental evaluation using two large benchmarks and six real-world data archives, we found ClaSS to be significantly more precise than eight state-of-the-art competitors. Its space and time complexity is independent of segment sizes and linear only in the sliding window size. We also provide ClaSS as a window operator with an average throughput of 538 data points per second for the Apache Flink streaming engine.
BELB: a Biomedical Entity Linking Benchmark
Aug 22, 2023



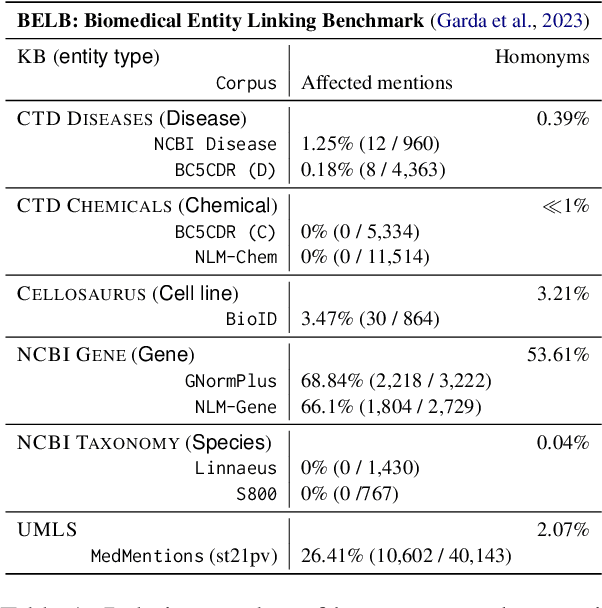
Biomedical entity linking (BEL) is the task of grounding entity mentions to a knowledge base. It plays a vital role in information extraction pipelines for the life sciences literature. We review recent work in the field and find that, as the task is absent from existing benchmarks for biomedical text mining, different studies adopt different experimental setups making comparisons based on published numbers problematic. Furthermore, neural systems are tested primarily on instances linked to the broad coverage knowledge base UMLS, leaving their performance to more specialized ones, e.g. genes or variants, understudied. We therefore developed BELB, a Biomedical Entity Linking Benchmark, providing access in a unified format to 11 corpora linked to 7 knowledge bases and spanning six entity types: gene, disease, chemical, species, cell line and variant. BELB greatly reduces preprocessing overhead in testing BEL systems on multiple corpora offering a standardized testbed for reproducible experiments. Using BELB we perform an extensive evaluation of six rule-based entity-specific systems and three recent neural approaches leveraging pre-trained language models. Our results reveal a mixed picture showing that neural approaches fail to perform consistently across entity types, highlighting the need of further studies towards entity-agnostic models.