 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeGeometry-First Generative Spatial Single-Cell Reconstruction
May 27, 2026Single-cell RNA sequencing (scRNA-seq) profiles large numbers of cells but loses spatial context, whereas spatial transcriptomics (ST) preserves partial spatial structure at lower resolution. Most existing integration methods either deconvolve spot mixtures or map cells onto a measured spot lattice, which ties reconstructions to a fixed grid and slide-specific coordinate systems, a limitation that is especially problematic in unpaired settings. We propose GEARS, a geometry-first framework that reconstructs an intrinsic single-cell spatial geometry guided by ST, without relying on cell-type labels, histological images, or cell-to-spot assignment. GEARS first learns a domain-invariant expression encoder that aligns ST spots and dissociated cells, and then trains a permutation-equivariant generator with a diffusion-based refiner with EDM-style preconditioning to generate local spatial geometries under pose-invariant supervision derived from ST coordinates. At inference, GEARS reconstructs geometry on many overlapping subsets of scRNA-seq cells, aggregates predicted pairwise distances across subsets, and solves a global distance-geometry problem to obtain canonical two-dimensional coordinates and a dense distance matrix. Extensive quantitative and qualitative experiments, including cross-section generalization, show that GEARS consistently improves global distance preservation, local neighborhood fidelity, and spatial distribution alignment compared to strong spatial mapping and deconvolution baselines.
Biological Pathway Guided Gene Selection Through Collaborative Reinforcement Learning
May 30, 2025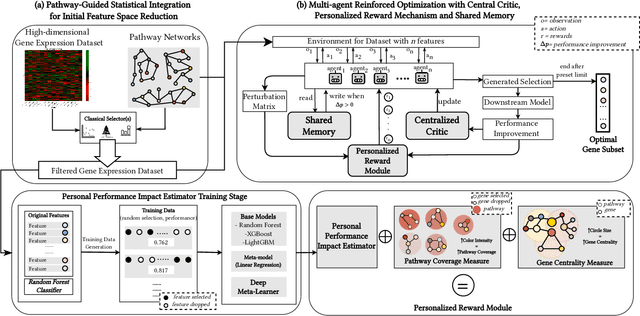
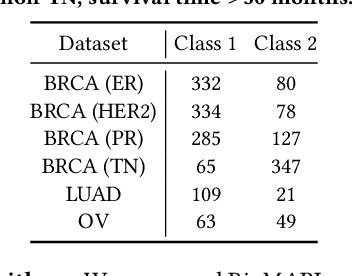
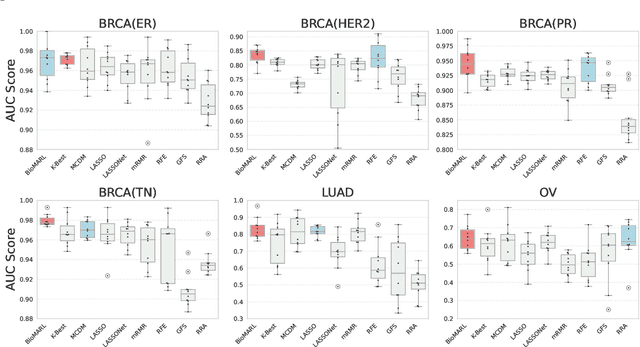
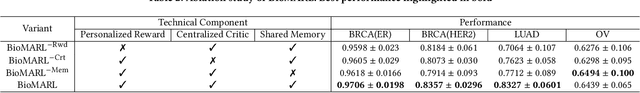
Gene selection in high-dimensional genomic data is essential for understanding disease mechanisms and improving therapeutic outcomes. Traditional feature selection methods effectively identify predictive genes but often ignore complex biological pathways and regulatory networks, leading to unstable and biologically irrelevant signatures. Prior approaches, such as Lasso-based methods and statistical filtering, either focus solely on individual gene-outcome associations or fail to capture pathway-level interactions, presenting a key challenge: how to integrate biological pathway knowledge while maintaining statistical rigor in gene selection? To address this gap, we propose a novel two-stage framework that integrates statistical selection with biological pathway knowledge using multi-agent reinforcement learning (MARL). First, we introduce a pathway-guided pre-filtering strategy that leverages multiple statistical methods alongside KEGG pathway information for initial dimensionality reduction. Next, for refined selection, we model genes as collaborative agents in a MARL framework, where each agent optimizes both predictive power and biological relevance. Our framework incorporates pathway knowledge through Graph Neural Network-based state representations, a reward mechanism combining prediction performance with gene centrality and pathway coverage, and collaborative learning strategies using shared memory and a centralized critic component. Extensive experiments on multiple gene expression datasets demonstrate that our approach significantly improves both prediction accuracy and biological interpretability compared to traditional methods.
Interaction of a priori Anatomic Knowledge with Self-Supervised Contrastive Learning in Cardiac Magnetic Resonance Imaging
May 25, 2022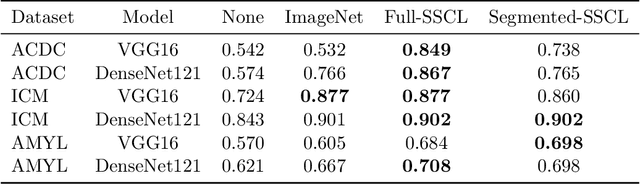
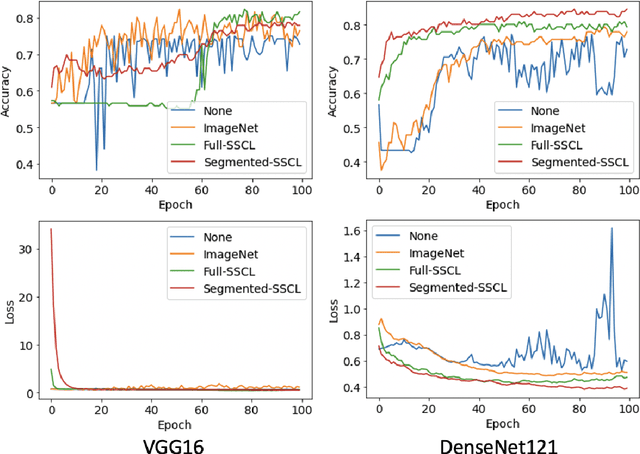
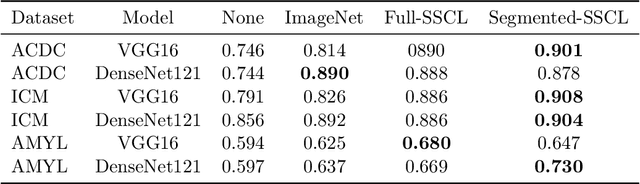
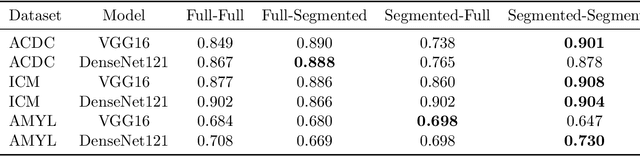
Training deep learning models on cardiac magnetic resonance imaging (CMR) can be a challenge due to the small amount of expert generated labels and inherent complexity of data source. Self-supervised contrastive learning (SSCL) has recently been shown to boost performance in several medical imaging tasks. However, it is unclear how much the pre-trained representation reflects the primary organ of interest compared to spurious surrounding tissue. In this work, we evaluate the optimal method of incorporating prior knowledge of anatomy into a SSCL training paradigm. Specifically, we evaluate using a segmentation network to explicitly local the heart in CMR images, followed by SSCL pretraining in multiple diagnostic tasks. We find that using a priori knowledge of anatomy can greatly improve the downstream diagnostic performance. Furthermore, SSCL pre-training with in-domain data generally improved downstream performance and more human-like saliency compared to end-to-end training and ImageNet pre-trained networks. However, introducing anatomic knowledge to pre-training generally does not have significant impact.
Automatic Skin Lesion Segmentation Using Deep Fully Convolutional Networks
Jul 17, 2018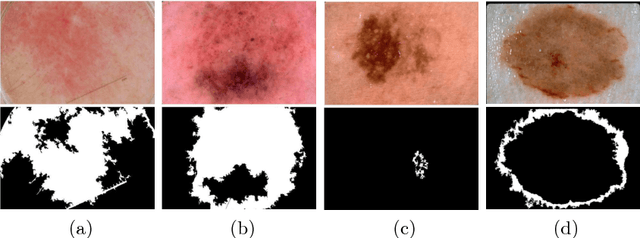

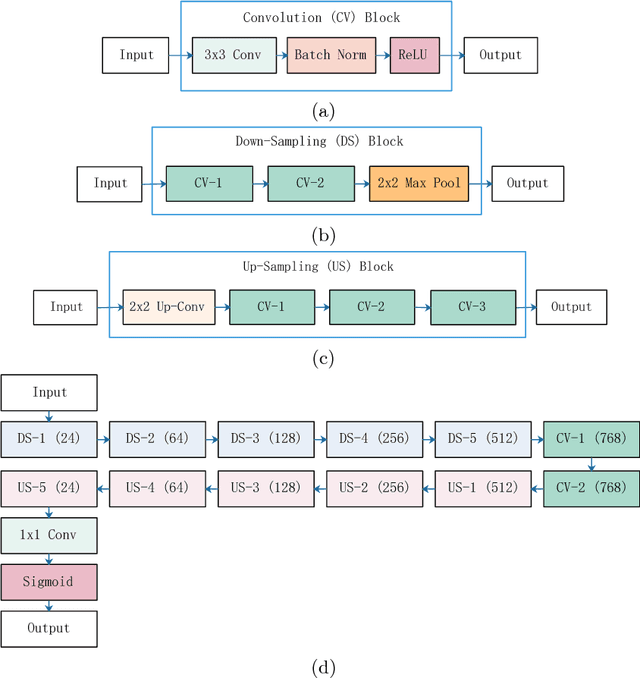
This paper summarizes our method and validation results for the ISIC Challenge 2018 - Skin Lesion Analysis Towards Melanoma Detection - Task 1: Lesion Segmentation
Bayesian Semi-nonnegative Tri-matrix Factorization to Identify Pathways Associated with Cancer Types
Dec 01, 2017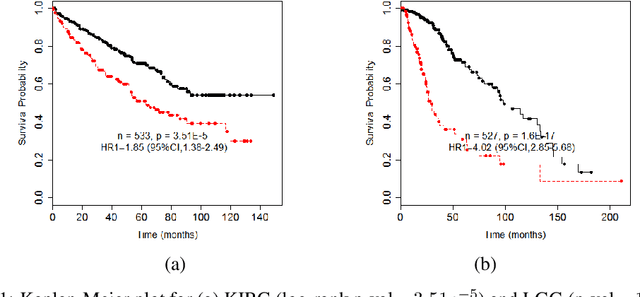
Identifying altered pathways that are associated with specific cancer types can potentially bring a significant impact on cancer patient treatment. Accurate identification of such key altered pathways information can be used to develop novel therapeutic agents as well as to understand the molecular mechanisms of various types of cancers better. Tri-matrix factorization is an efficient tool to learn associations between two different entities (e.g., cancer types and pathways in our case) from data. To successfully apply tri-matrix factorization methods to biomedical problems, biological prior knowledge such as pathway databases or protein-protein interaction (PPI) networks, should be taken into account in the factorization model. However, it is not straightforward in the Bayesian setting even though Bayesian methods are more appealing than point estimate methods, such as a maximum likelihood or a maximum posterior method, in the sense that they calculate distributions over variables and are robust against overfitting. We propose a Bayesian (semi-)nonnegative matrix factorization model for human cancer genomic data, where the biological prior knowledge represented by a pathway database and a PPI network is taken into account in the factorization model through a finite dependent Beta-Bernoulli prior. We tested our method on The Cancer Genome Atlas (TCGA) dataset and found that the pathways identified by our method can be used as a prognostic biomarkers for patient subgroup identification.