 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgemTim: Rapid and accurate transcript reconstruction from RNA-Seq data
Sep 20, 2013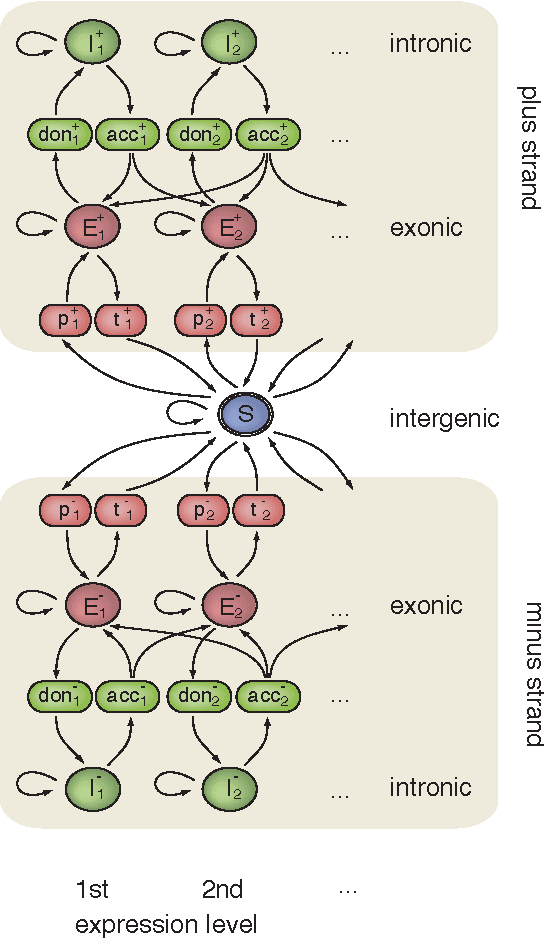
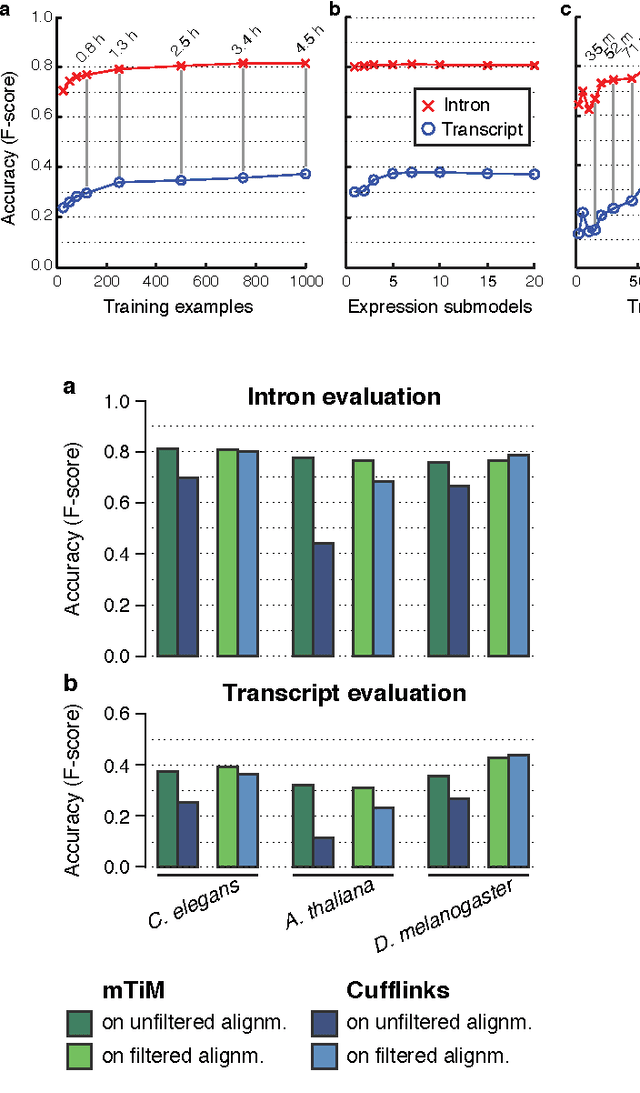
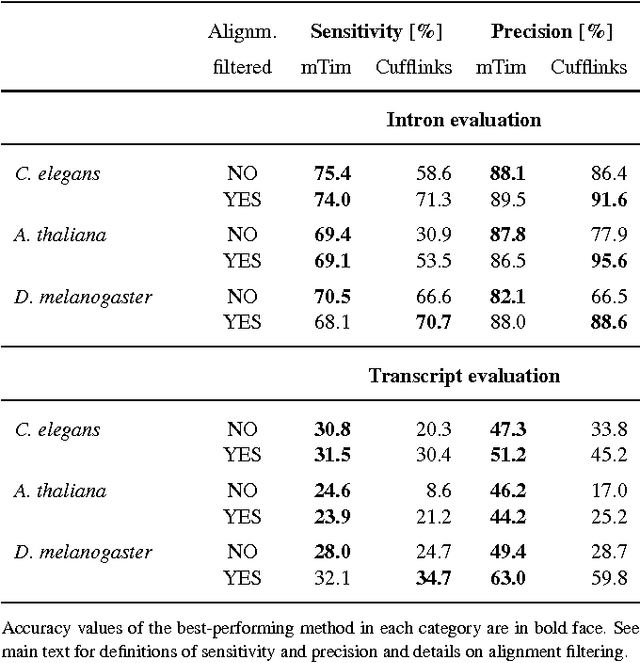
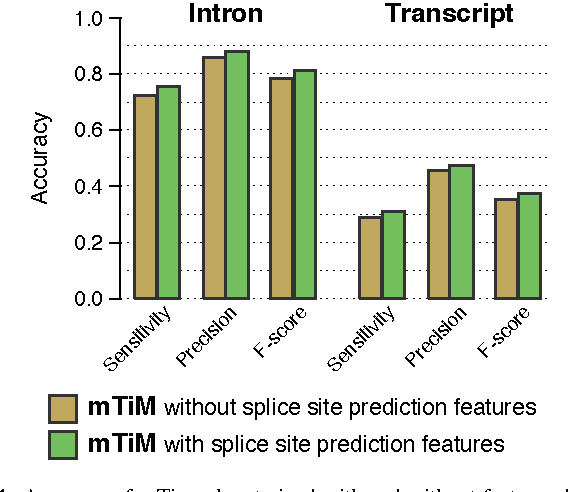
Recent advances in high-throughput cDNA sequencing (RNA-Seq) technology have revolutionized transcriptome studies. A major motivation for RNA-Seq is to map the structure of expressed transcripts at nucleotide resolution. With accurate computational tools for transcript reconstruction, this technology may also become useful for genome (re-)annotation, which has mostly relied on de novo gene finding where gene structures are primarily inferred from the genome sequence. We developed a machine-learning method, called mTim (margin-based transcript inference method) for transcript reconstruction from RNA-Seq read alignments that is based on discriminatively trained hidden Markov support vector machines. In addition to features derived from read alignments, it utilizes characteristic genomic sequences, e.g. around splice sites, to improve transcript predictions. mTim inferred transcripts that were highly accurate and relatively robust to alignment errors in comparison to those from Cufflinks, a widely used transcript assembly method.
Non-Sparse Regularization for Multiple Kernel Learning
Oct 26, 2010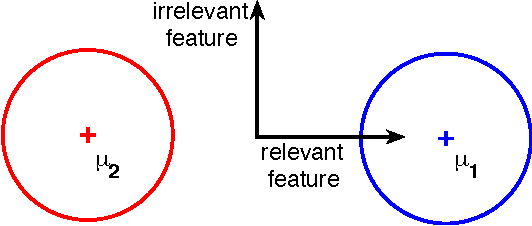
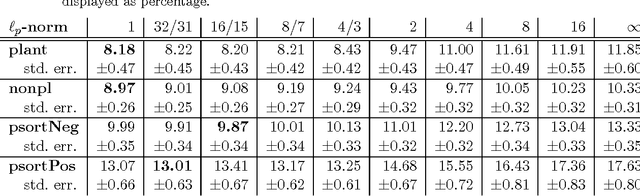
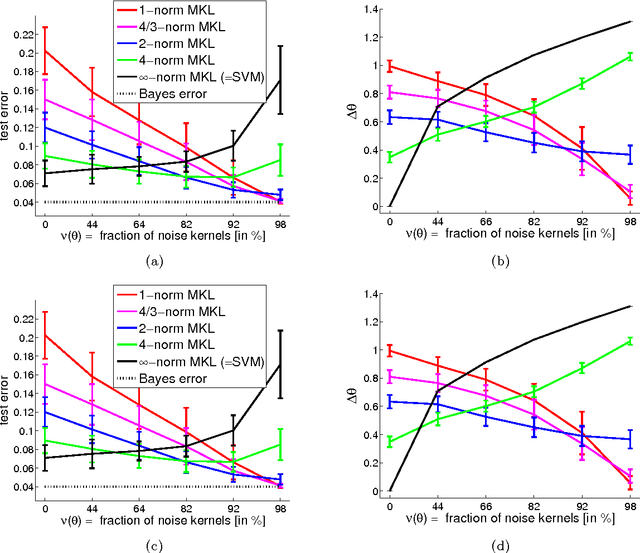
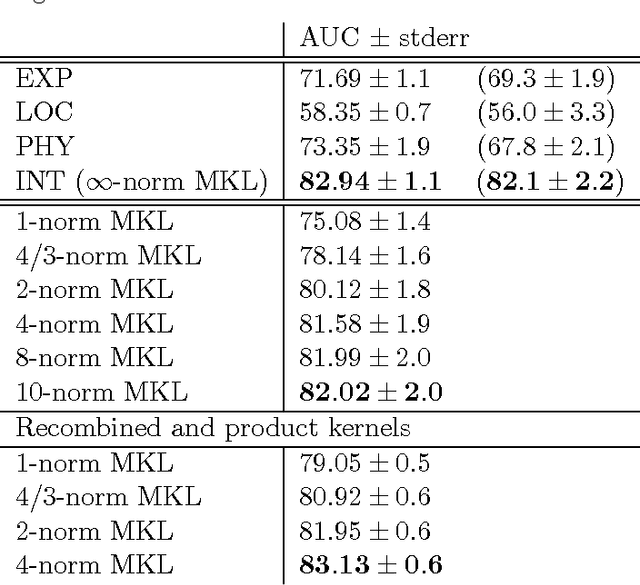
Learning linear combinations of multiple kernels is an appealing strategy when the right choice of features is unknown. Previous approaches to multiple kernel learning (MKL) promote sparse kernel combinations to support interpretability and scalability. Unfortunately, this 1-norm MKL is rarely observed to outperform trivial baselines in practical applications. To allow for robust kernel mixtures, we generalize MKL to arbitrary norms. We devise new insights on the connection between several existing MKL formulations and develop two efficient interleaved optimization strategies for arbitrary norms, like p-norms with p>1. Empirically, we demonstrate that the interleaved optimization strategies are much faster compared to the commonly used wrapper approaches. A theoretical analysis and an experiment on controlled artificial data experiment sheds light on the appropriateness of sparse, non-sparse and $\ell_\infty$-norm MKL in various scenarios. Empirical applications of p-norm MKL to three real-world problems from computational biology show that non-sparse MKL achieves accuracies that go beyond the state-of-the-art.
The Feature Importance Ranking Measure
Jun 23, 2009
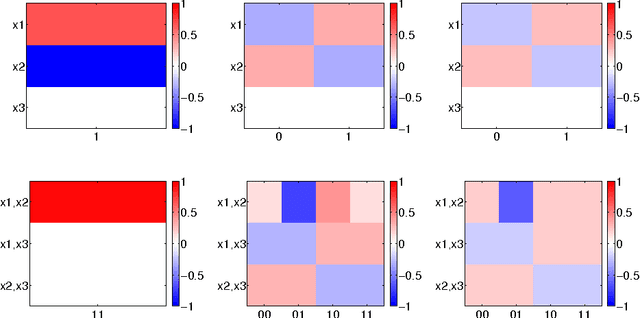
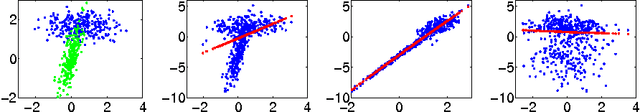
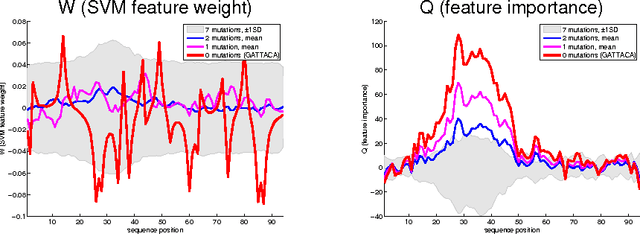
Most accurate predictions are typically obtained by learning machines with complex feature spaces (as e.g. induced by kernels). Unfortunately, such decision rules are hardly accessible to humans and cannot easily be used to gain insights about the application domain. Therefore, one often resorts to linear models in combination with variable selection, thereby sacrificing some predictive power for presumptive interpretability. Here, we introduce the Feature Importance Ranking Measure (FIRM), which by retrospective analysis of arbitrary learning machines allows to achieve both excellent predictive performance and superior interpretation. In contrast to standard raw feature weighting, FIRM takes the underlying correlation structure of the features into account. Thereby, it is able to discover the most relevant features, even if their appearance in the training data is entirely prevented by noise. The desirable properties of FIRM are investigated analytically and illustrated in simulations.
* 15 pages, 3 figures. to appear in the Proceedings of the European Conference on Machine Learning and Principles and Practice of Knowledge Discovery in Databases (ECML/PKDD), 2009