 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgemTim: Rapid and accurate transcript reconstruction from RNA-Seq data
Paper and Code
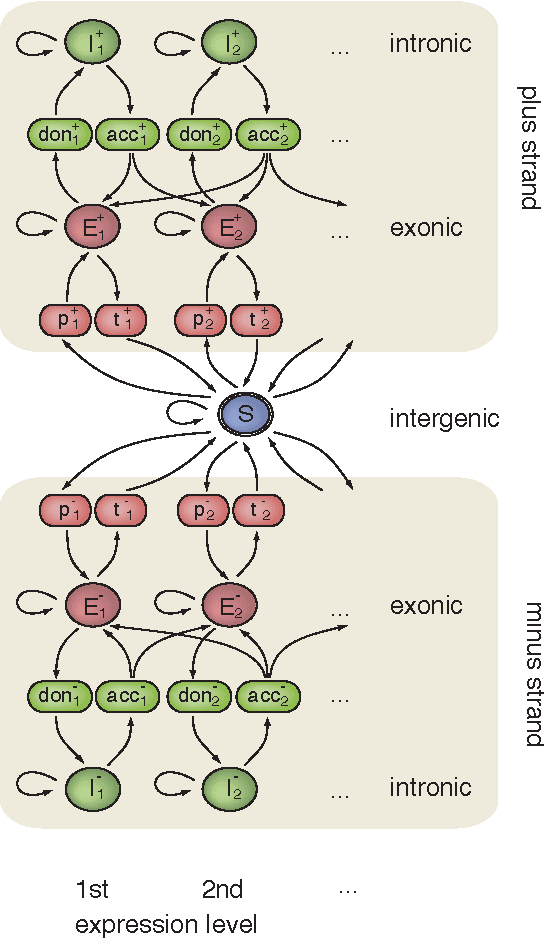
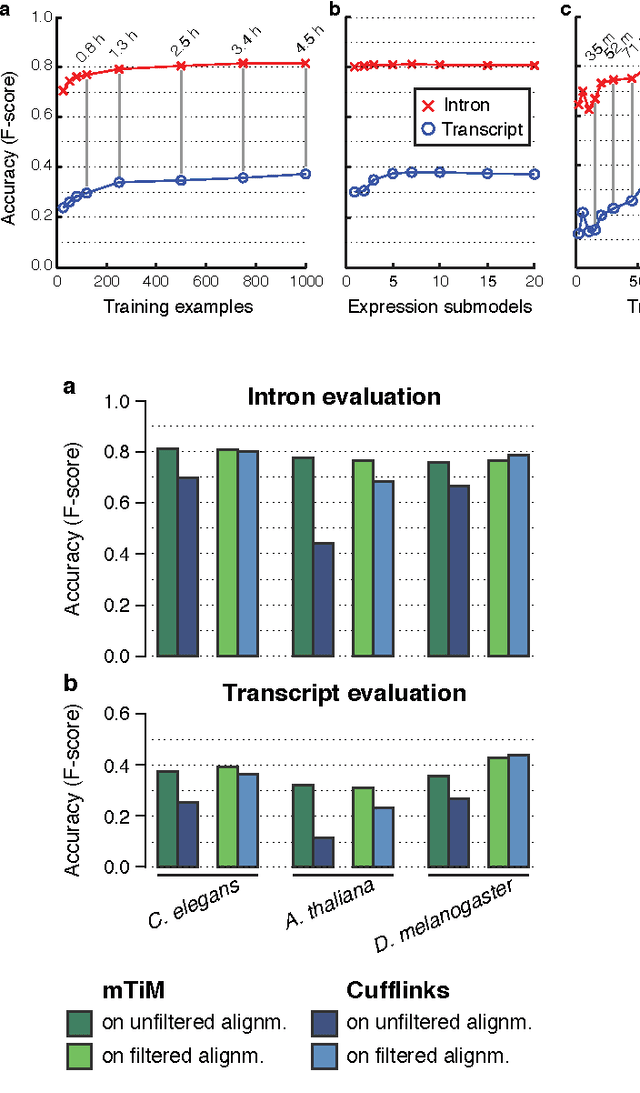
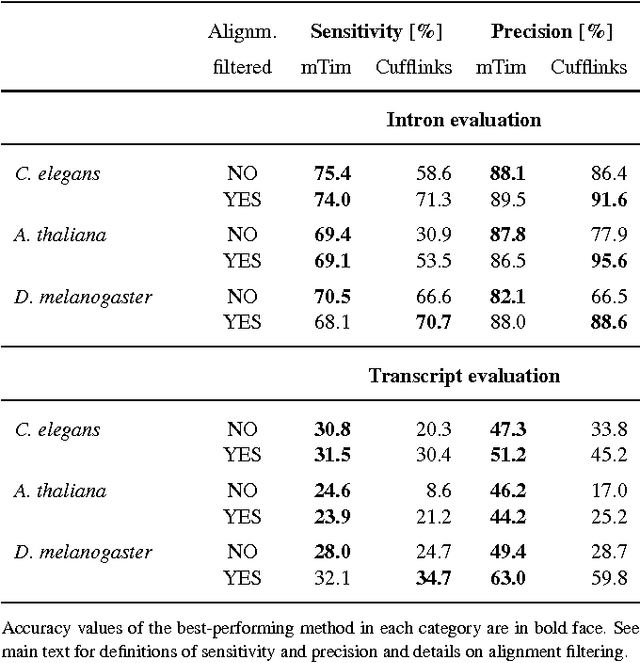
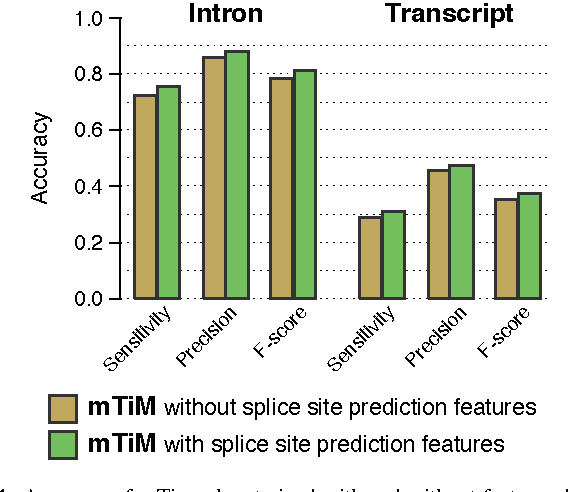
Recent advances in high-throughput cDNA sequencing (RNA-Seq) technology have revolutionized transcriptome studies. A major motivation for RNA-Seq is to map the structure of expressed transcripts at nucleotide resolution. With accurate computational tools for transcript reconstruction, this technology may also become useful for genome (re-)annotation, which has mostly relied on de novo gene finding where gene structures are primarily inferred from the genome sequence. We developed a machine-learning method, called mTim (margin-based transcript inference method) for transcript reconstruction from RNA-Seq read alignments that is based on discriminatively trained hidden Markov support vector machines. In addition to features derived from read alignments, it utilizes characteristic genomic sequences, e.g. around splice sites, to improve transcript predictions. mTim inferred transcripts that were highly accurate and relatively robust to alignment errors in comparison to those from Cufflinks, a widely used transcript assembly method.