 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeA practical guide to machine learning interatomic potentials -- Status and future
Mar 12, 2025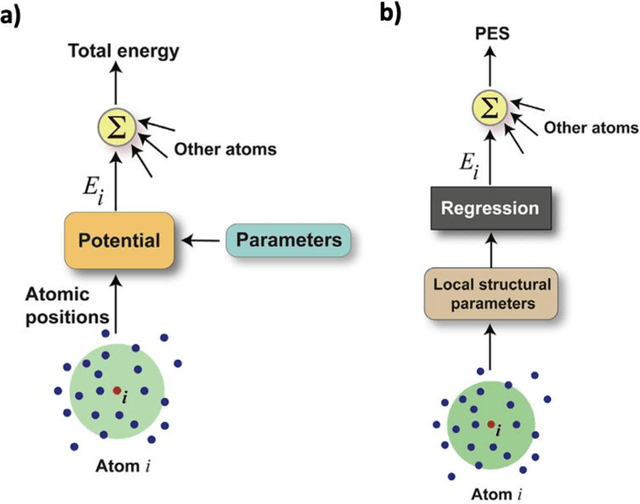

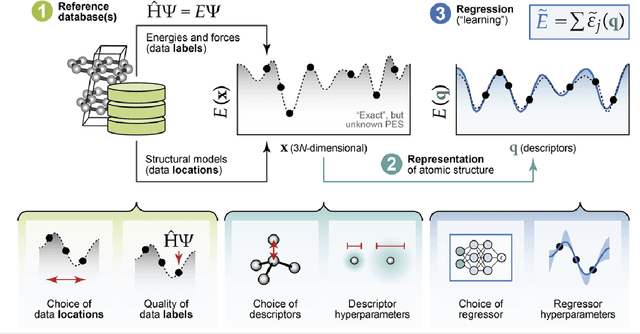
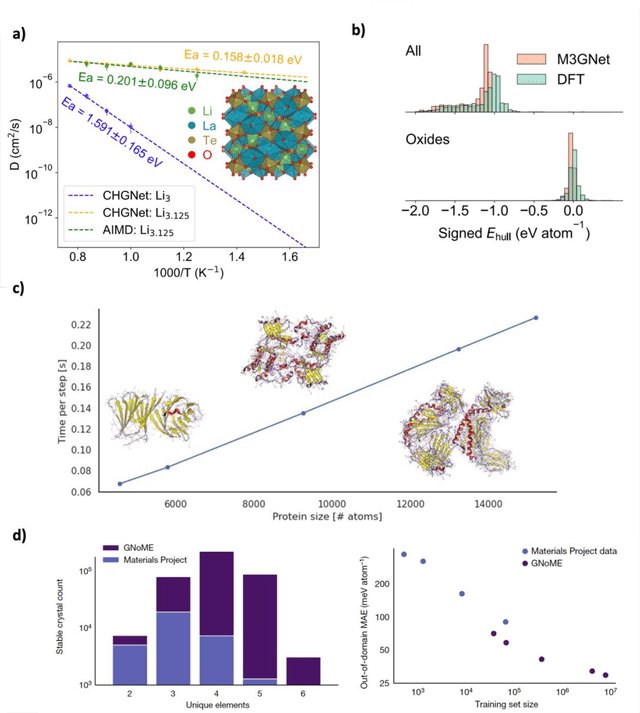
The rapid development and large body of literature on machine learning interatomic potentials (MLIPs) can make it difficult to know how to proceed for researchers who are not experts but wish to use these tools. The spirit of this review is to help such researchers by serving as a practical, accessible guide to the state-of-the-art in MLIPs. This review paper covers a broad range of topics related to MLIPs, including (i) central aspects of how and why MLIPs are enablers of many exciting advancements in molecular modeling, (ii) the main underpinnings of different types of MLIPs, including their basic structure and formalism, (iii) the potentially transformative impact of universal MLIPs for both organic and inorganic systems, including an overview of the most recent advances, capabilities, downsides, and potential applications of this nascent class of MLIPs, (iv) a practical guide for estimating and understanding the execution speed of MLIPs, including guidance for users based on hardware availability, type of MLIP used, and prospective simulation size and time, (v) a manual for what MLIP a user should choose for a given application by considering hardware resources, speed requirements, energy and force accuracy requirements, as well as guidance for choosing pre-trained potentials or fitting a new potential from scratch, (vi) discussion around MLIP infrastructure, including sources of training data, pre-trained potentials, and hardware resources for training, (vii) summary of some key limitations of present MLIPs and current approaches to mitigate such limitations, including methods of including long-range interactions, handling magnetic systems, and treatment of excited states, and finally (viii) we finish with some more speculative thoughts on what the future holds for the development and application of MLIPs over the next 3-10+ years.
Generative Model for Constructing Reaction Path from Initial to Final States
Jan 19, 2024
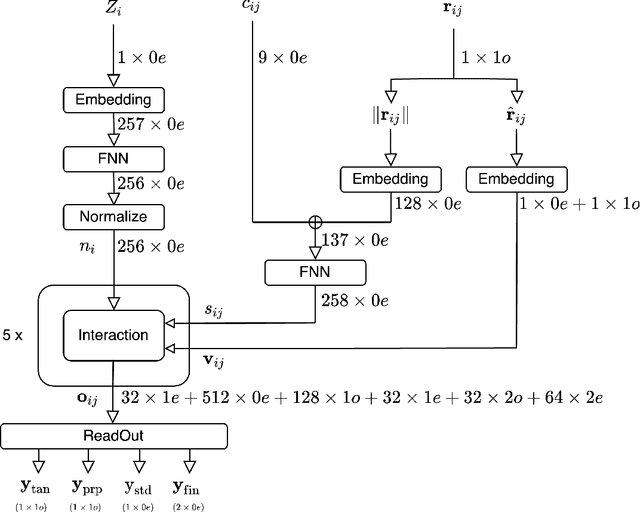
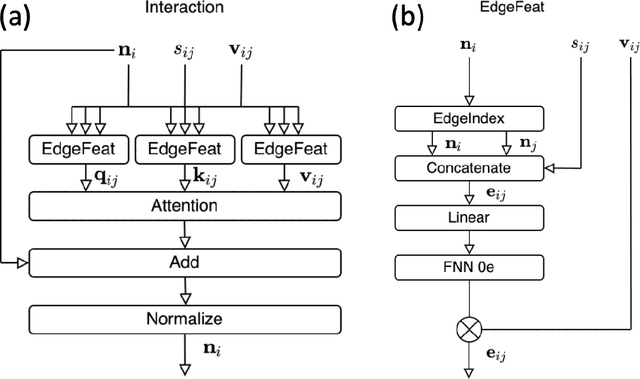
Mapping out reaction pathways and their corresponding activation barriers is a significant aspect of molecular simulation. Given their inherent complexity and nonlinearity, even generating a initial guess of these paths remains a challenging problem. Presented in this paper is an innovative approach that utilizes neural networks to generate initial guess for these reaction pathways. The proposed method is initiated by inputting the coordinates of the initial state, followed by progressive alterations to its structure. This iterative process culminates in the generation of the approximate representation of the reaction path and the coordinates of the final state. The application of this method extends to complex reaction pathways illustrated by organic reactions. Training was executed on the Transition1x dataset, an organic reaction pathway dataset. The results revealed generation of reactions that bore substantial similarities with the corresponding test data. The method's flexibility allows for reactions to be generated either to conform to predetermined conditions or in a randomized manner.
TeaNet: universal neural network interatomic potential inspired by iterative electronic relaxations
Dec 02, 2019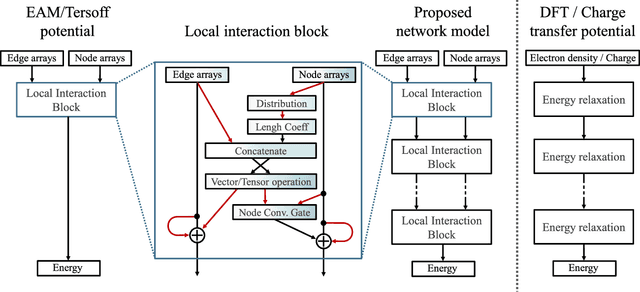
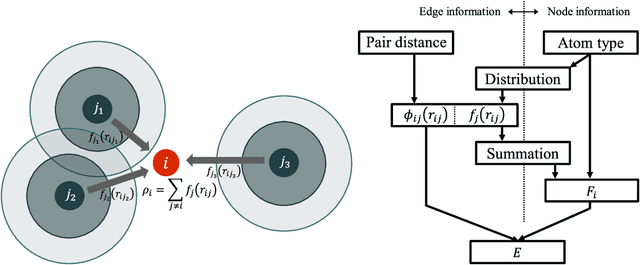
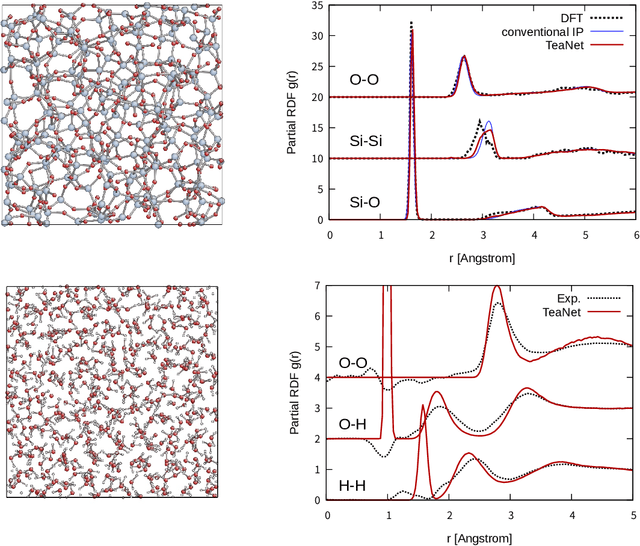
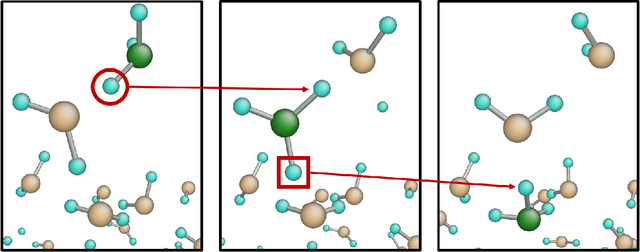
A universal interatomic potential applicable to arbitrary elements and structures is urgently needed in computational materials science. Graph convolution-based neural network is a promising approach by virtue of its ability to express complex relations. Thus far, it has been thought to represent a completely different approach from physics-based interatomic potentials. In this paper, we show that these two methods can be regarded as different representations of the same tight-binding electronic relaxation framework, where atom-based and overlap integral or "bond"-based Hamiltonian information are propagated in a directional fashion. Based on this unified view, we propose a new model, named the tensor embedded atom network (TeaNet), where the stacked network model is associated with the electronic total energy relaxation calculation. Furthermore, Tersoff-style angular interaction is translated into graph convolution architecture through the incorporation of Euclidean tensor values. Our model can represent and transfer spatial information. TeaNet shows great performance in both the robustness of interatomic potentials and the expressive power of neural networks. We demonstrate that arbitrary chemistry involving the first 18 elements on the periodic table (H to Ar) can be realized by our model, including C-H molecular structures, metals, amorphous SiO${}_2$, and water.