 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeGenerative Model for Constructing Reaction Path from Initial to Final States
Jan 19, 2024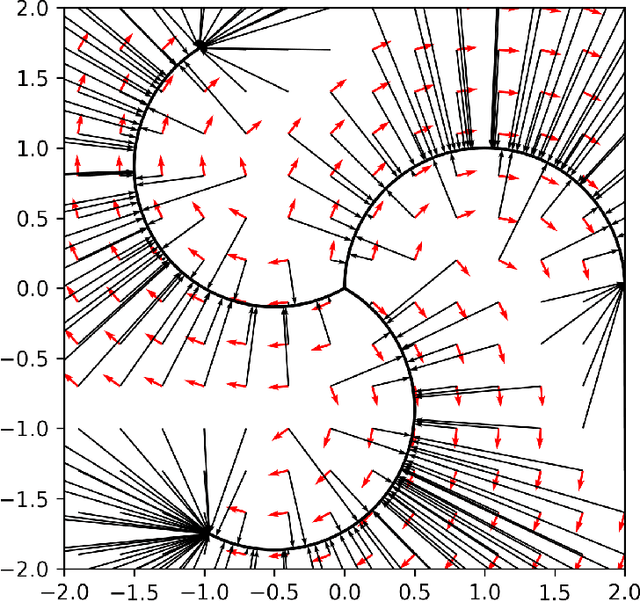
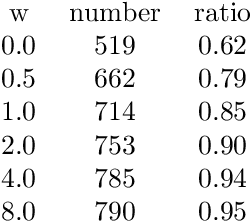
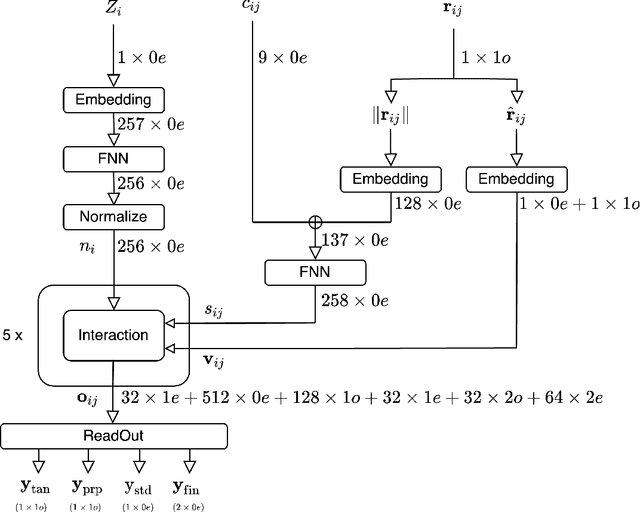
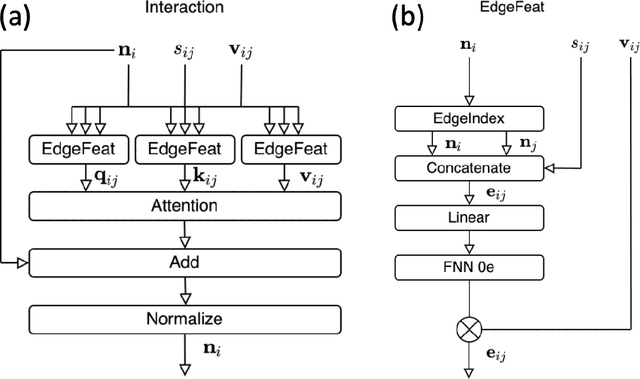
Mapping out reaction pathways and their corresponding activation barriers is a significant aspect of molecular simulation. Given their inherent complexity and nonlinearity, even generating a initial guess of these paths remains a challenging problem. Presented in this paper is an innovative approach that utilizes neural networks to generate initial guess for these reaction pathways. The proposed method is initiated by inputting the coordinates of the initial state, followed by progressive alterations to its structure. This iterative process culminates in the generation of the approximate representation of the reaction path and the coordinates of the final state. The application of this method extends to complex reaction pathways illustrated by organic reactions. Training was executed on the Transition1x dataset, an organic reaction pathway dataset. The results revealed generation of reactions that bore substantial similarities with the corresponding test data. The method's flexibility allows for reactions to be generated either to conform to predetermined conditions or in a randomized manner.