 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeIn-Pocket 3D Graphs Enhance Ligand-Target Compatibility in Generative Small-Molecule Creation
Apr 05, 2022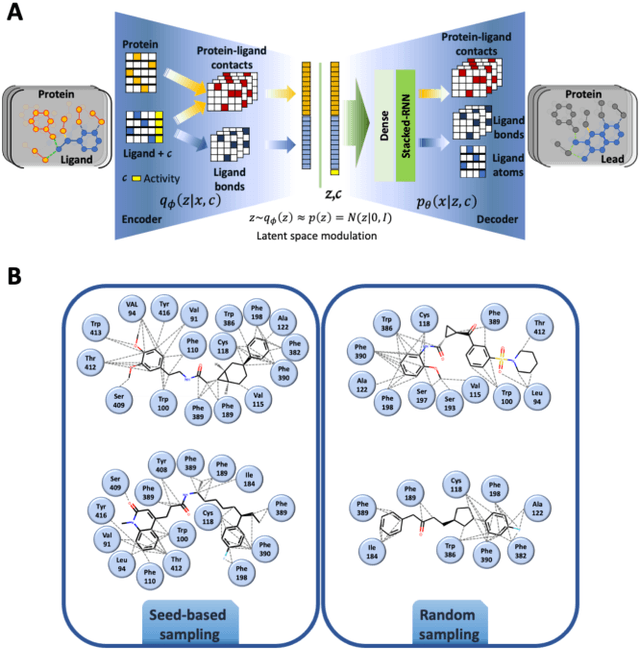

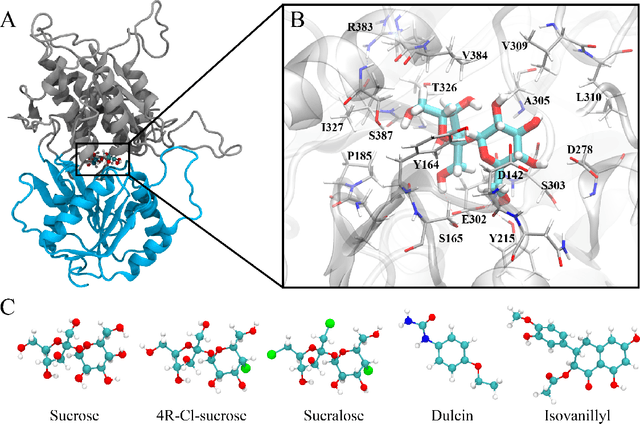
Proteins in complex with small molecule ligands represent the core of structure-based drug discovery. However, three-dimensional representations are absent from most deep-learning-based generative models. We here present a graph-based generative modeling technology that encodes explicit 3D protein-ligand contacts within a relational graph architecture. The models combine a conditional variational autoencoder that allows for activity-specific molecule generation with putative contact generation that provides predictions of molecular interactions within the target binding pocket. We show that molecules generated with our 3D procedure are more compatible with the binding pocket of the dopamine D2 receptor than those produced by a comparable ligand-based 2D generative method, as measured by docking scores, expected stereochemistry, and recoverability in commercial chemical databases. Predicted protein-ligand contacts were found among highest-ranked docking poses with a high recovery rate. This work shows how the structural context of a protein target can be used to enhance molecule generation.
Analysis of training and seed bias in small molecules generated with a conditional graph-based variational autoencoder -- Insights for practical AI-driven molecule generation
Jul 19, 2021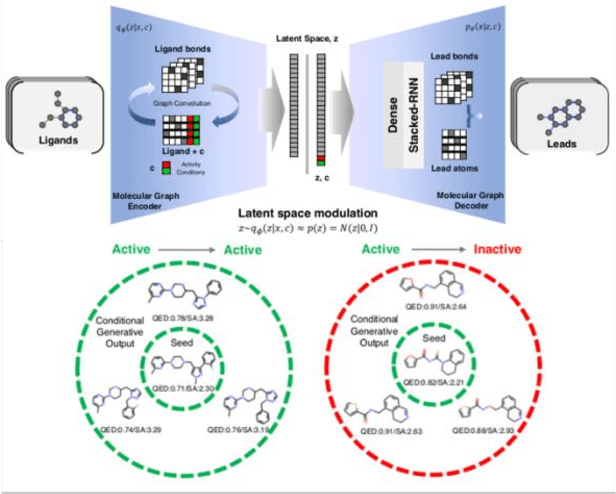
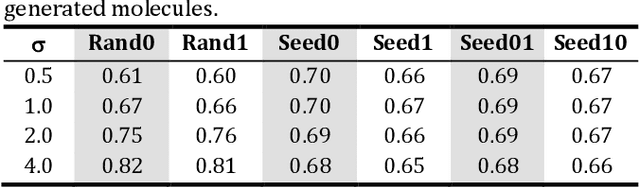
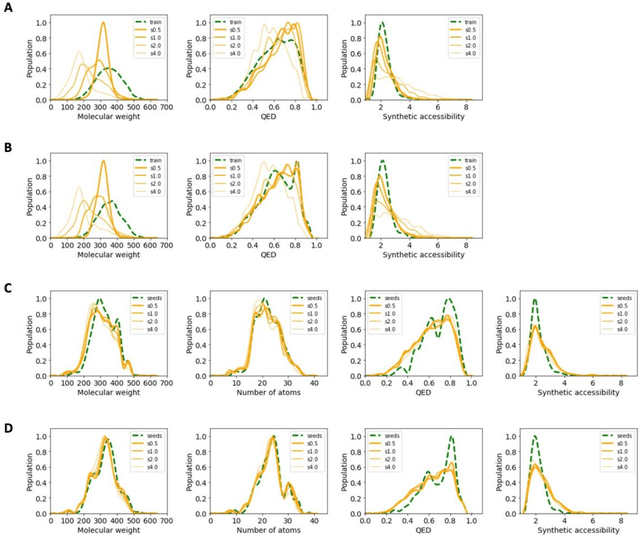
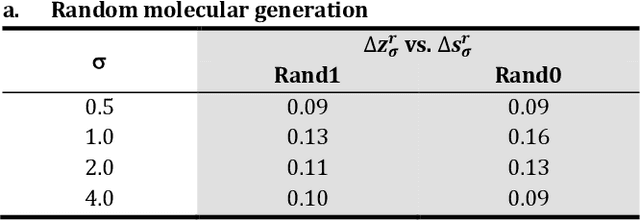
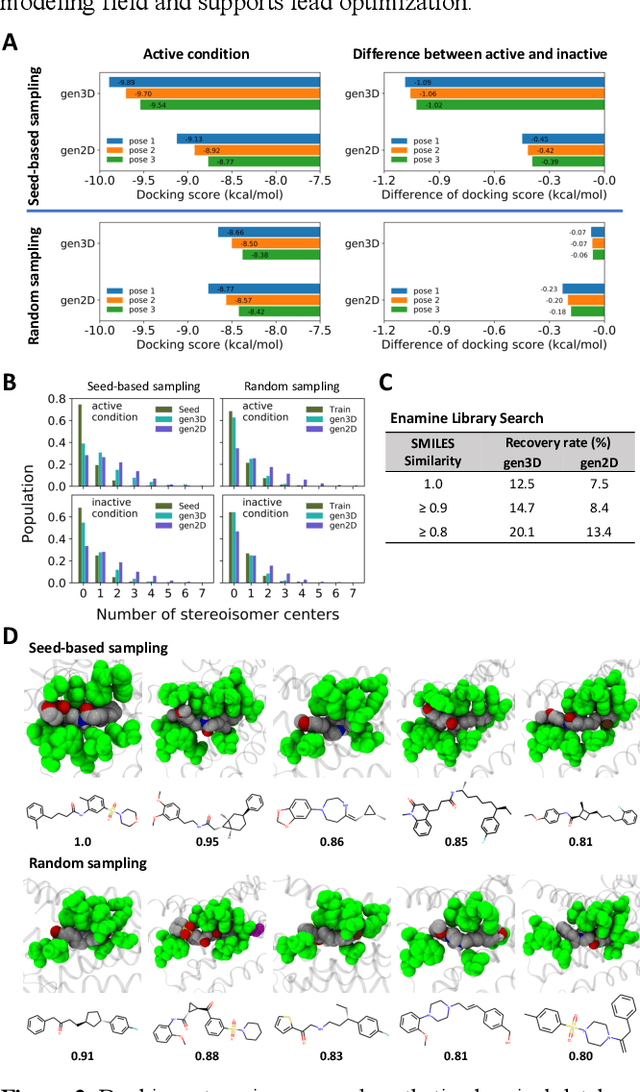
The application of deep learning to generative molecule design has shown early promise for accelerating lead series development. However, questions remain concerning how factors like training, dataset, and seed bias impact the technology's utility to medicine and computational chemists. In this work, we analyze the impact of seed and training bias on the output of an activity-conditioned graph-based variational autoencoder (VAE). Leveraging a massive, labeled dataset corresponding to the dopamine D2 receptor, our graph-based generative model is shown to excel in producing desired conditioned activities and favorable unconditioned physical properties in generated molecules. We implement an activity swapping method that allows for the activation, deactivation, or retention of activity of molecular seeds, and we apply independent deep learning classifiers to verify the generative results. Overall, we uncover relationships between noise, molecular seeds, and training set selection across a range of latent-space sampling procedures, providing important insights for practical AI-driven molecule generation.
CASTELO: Clustered Atom Subtypes aidEd Lead Optimization -- a combined machine learning and molecular modeling method
Nov 27, 2020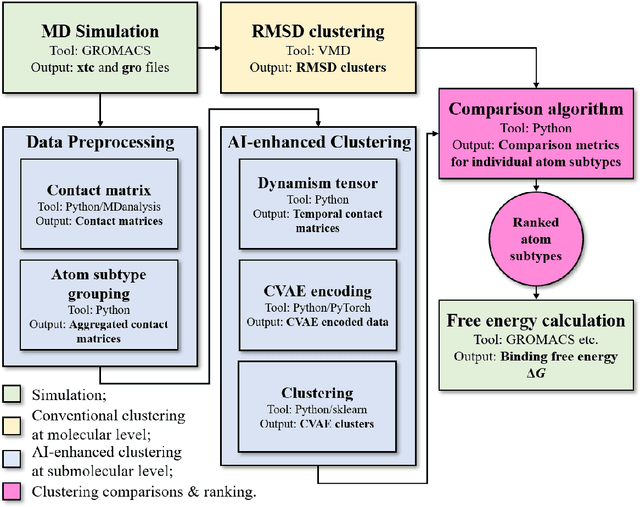

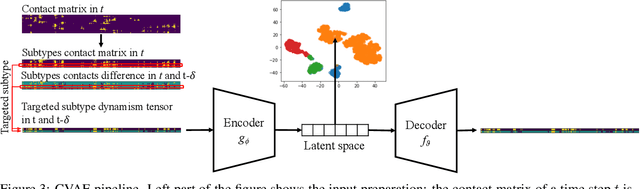
Drug discovery is a multi-stage process that comprises two costly major steps: pre-clinical research and clinical trials. Among its stages, lead optimization easily consumes more than half of the pre-clinical budget. We propose a combined machine learning and molecular modeling approach that automates lead optimization workflow \textit{in silico}. The initial data collection is achieved with physics-based molecular dynamics (MD) simulation. Contact matrices are calculated as the preliminary features extracted from the simulations. To take advantage of the temporal information from the simulations, we enhanced contact matrices data with temporal dynamism representation, which are then modeled with unsupervised convolutional variational autoencoder (CVAE). Finally, conventional clustering method and CVAE-based clustering method are compared with metrics to rank the submolecular structures and propose potential candidates for lead optimization. With no need for extensive structure-activity relationship database, our method provides new hints for drug modification hotspots which can be used to improve drug efficacy. Our workflow can potentially reduce the lead optimization turnaround time from months/years to days compared with the conventional labor-intensive process and thus can potentially become a valuable tool for medical researchers.