 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeCASTELO: Clustered Atom Subtypes aidEd Lead Optimization -- a combined machine learning and molecular modeling method
Paper and Code
Nov 27, 2020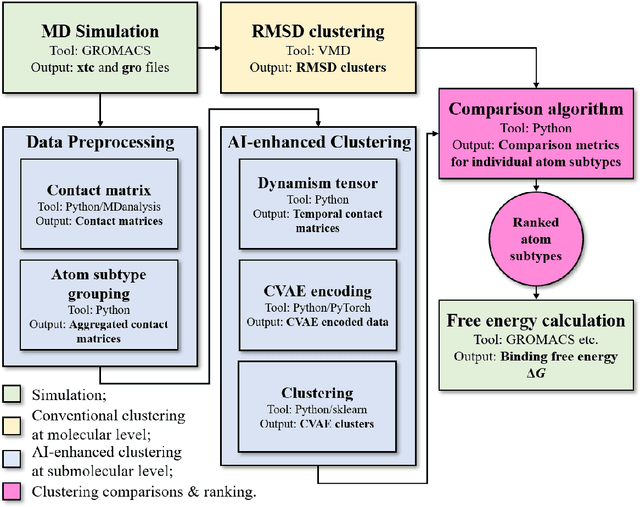

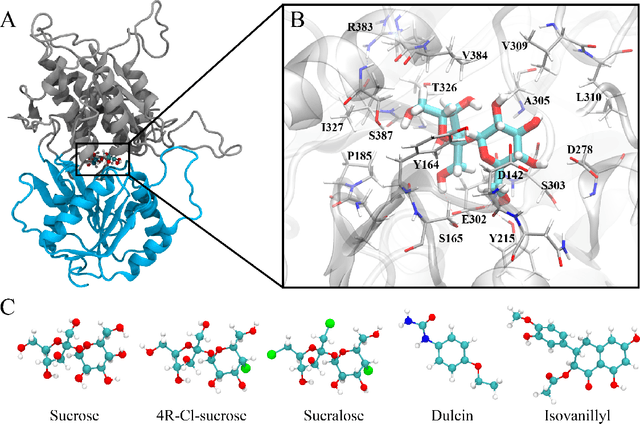
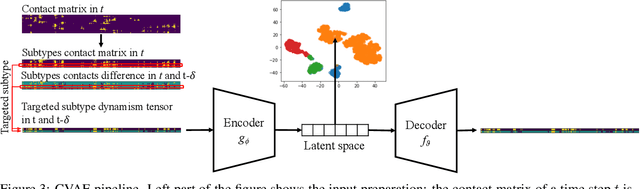
Drug discovery is a multi-stage process that comprises two costly major steps: pre-clinical research and clinical trials. Among its stages, lead optimization easily consumes more than half of the pre-clinical budget. We propose a combined machine learning and molecular modeling approach that automates lead optimization workflow \textit{in silico}. The initial data collection is achieved with physics-based molecular dynamics (MD) simulation. Contact matrices are calculated as the preliminary features extracted from the simulations. To take advantage of the temporal information from the simulations, we enhanced contact matrices data with temporal dynamism representation, which are then modeled with unsupervised convolutional variational autoencoder (CVAE). Finally, conventional clustering method and CVAE-based clustering method are compared with metrics to rank the submolecular structures and propose potential candidates for lead optimization. With no need for extensive structure-activity relationship database, our method provides new hints for drug modification hotspots which can be used to improve drug efficacy. Our workflow can potentially reduce the lead optimization turnaround time from months/years to days compared with the conventional labor-intensive process and thus can potentially become a valuable tool for medical researchers.