 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeIn-Pocket 3D Graphs Enhance Ligand-Target Compatibility in Generative Small-Molecule Creation
Apr 05, 2022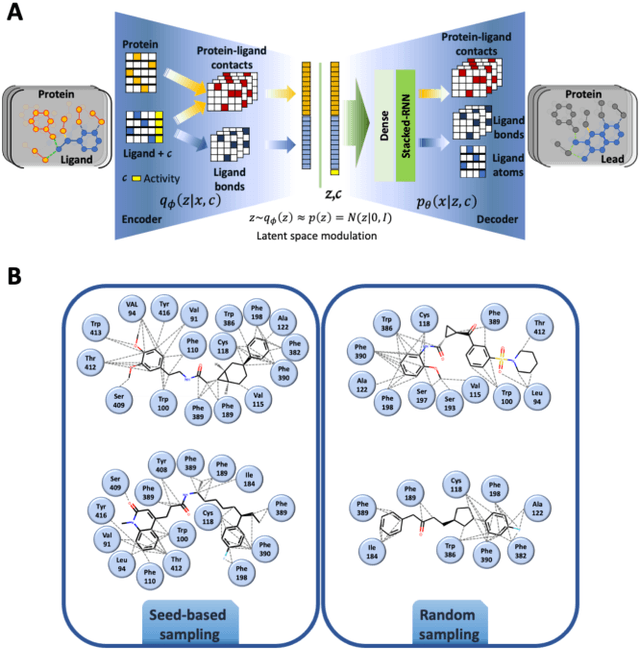

Proteins in complex with small molecule ligands represent the core of structure-based drug discovery. However, three-dimensional representations are absent from most deep-learning-based generative models. We here present a graph-based generative modeling technology that encodes explicit 3D protein-ligand contacts within a relational graph architecture. The models combine a conditional variational autoencoder that allows for activity-specific molecule generation with putative contact generation that provides predictions of molecular interactions within the target binding pocket. We show that molecules generated with our 3D procedure are more compatible with the binding pocket of the dopamine D2 receptor than those produced by a comparable ligand-based 2D generative method, as measured by docking scores, expected stereochemistry, and recoverability in commercial chemical databases. Predicted protein-ligand contacts were found among highest-ranked docking poses with a high recovery rate. This work shows how the structural context of a protein target can be used to enhance molecule generation.
Analysis of training and seed bias in small molecules generated with a conditional graph-based variational autoencoder -- Insights for practical AI-driven molecule generation
Jul 19, 2021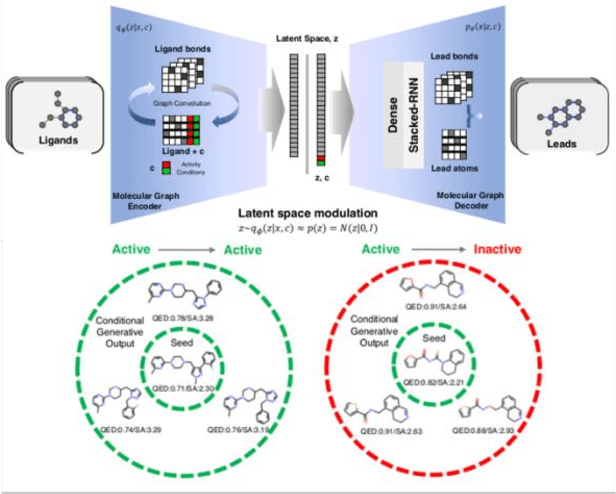
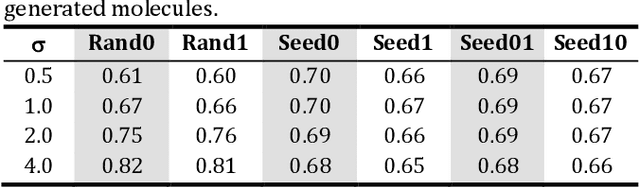
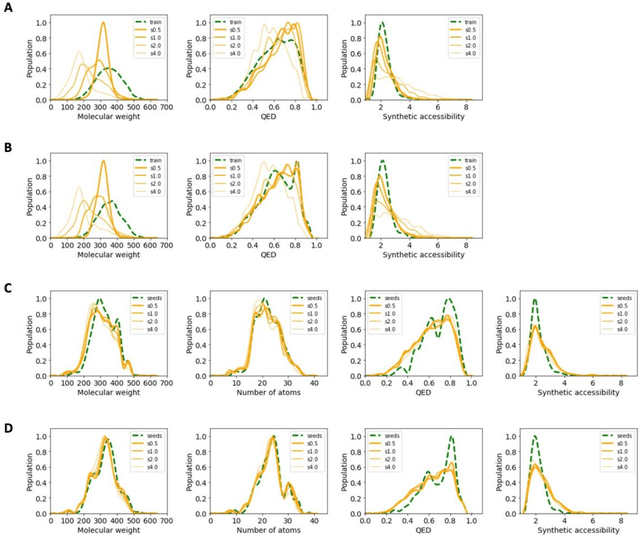
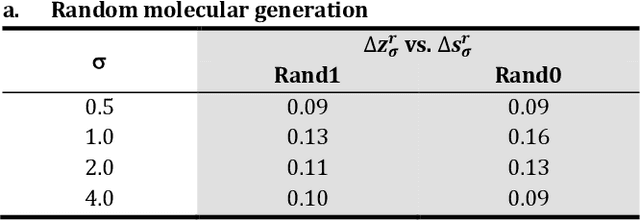
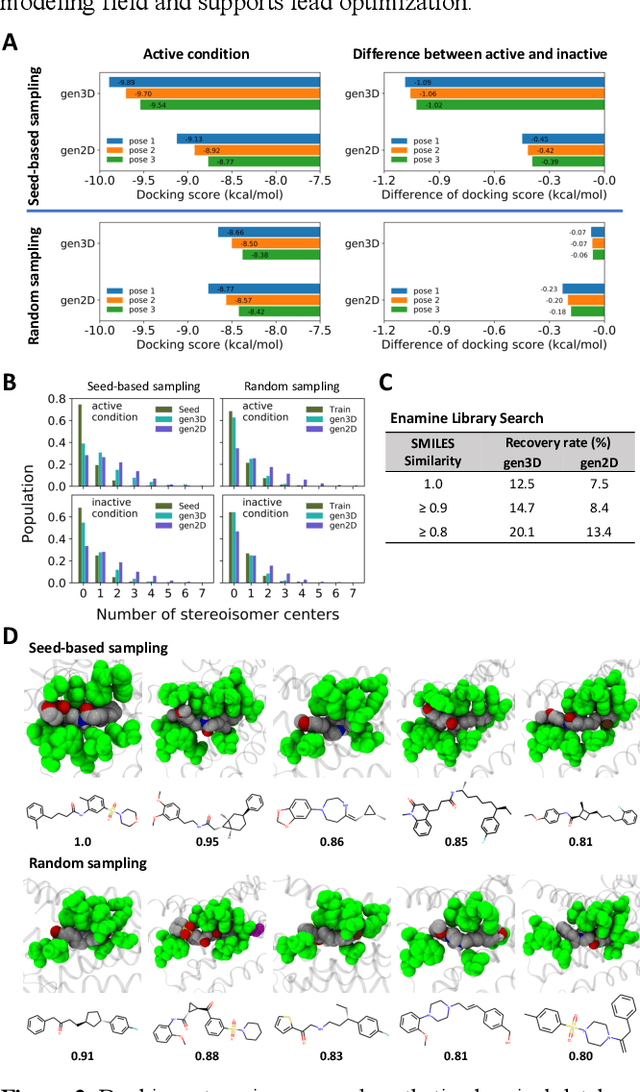
The application of deep learning to generative molecule design has shown early promise for accelerating lead series development. However, questions remain concerning how factors like training, dataset, and seed bias impact the technology's utility to medicine and computational chemists. In this work, we analyze the impact of seed and training bias on the output of an activity-conditioned graph-based variational autoencoder (VAE). Leveraging a massive, labeled dataset corresponding to the dopamine D2 receptor, our graph-based generative model is shown to excel in producing desired conditioned activities and favorable unconditioned physical properties in generated molecules. We implement an activity swapping method that allows for the activation, deactivation, or retention of activity of molecular seeds, and we apply independent deep learning classifiers to verify the generative results. Overall, we uncover relationships between noise, molecular seeds, and training set selection across a range of latent-space sampling procedures, providing important insights for practical AI-driven molecule generation.
Combining docking pose rank and structure with deep learning improves protein-ligand binding mode prediction
Oct 07, 2019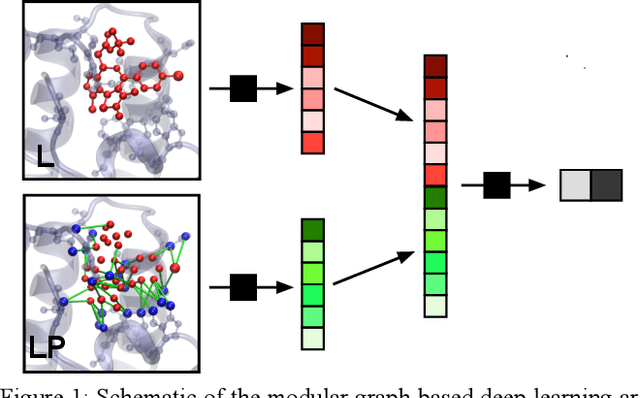
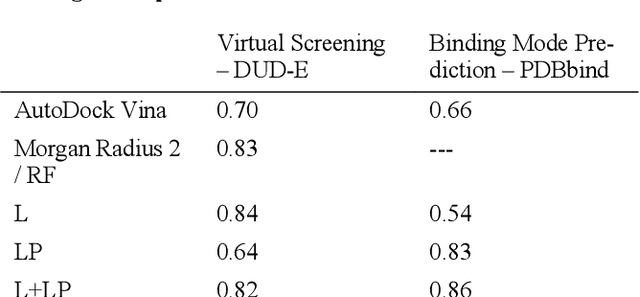
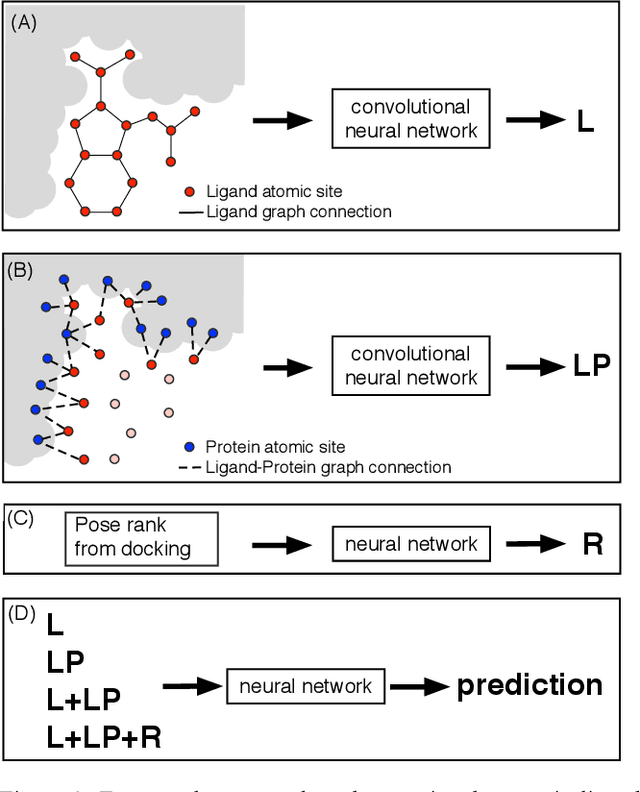
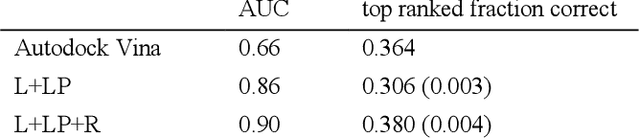
We present a simple, modular graph-based convolutional neural network that takes structural information from protein-ligand complexes as input to generate models for activity and binding mode prediction. Complex structures are generated by a standard docking procedure and fed into a dual-graph architecture that includes separate sub-networks for the ligand bonded topology and the ligand-protein contact map. This network division allows contributions from ligand identity to be distinguished from effects of protein-ligand interactions on classification. We show, in agreement with recent literature, that dataset bias drives many of the promising results on virtual screening that have previously been reported. However, we also show that our neural network is capable of learning from protein structural information when, as in the case of binding mode prediction, an unbiased dataset is constructed. We develop a deep learning model for binding mode prediction that uses docking ranking as input in combination with docking structures. This strategy mirrors past consensus models and outperforms the baseline docking program in a variety of tests, including on cross-docking datasets that mimic real-world docking use cases. Furthermore, the magnitudes of network predictions serve as reliable measures of model confidence