 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeScalable Label Propagation for Multi-relational Learning on Tensor Product Graph
Feb 20, 2018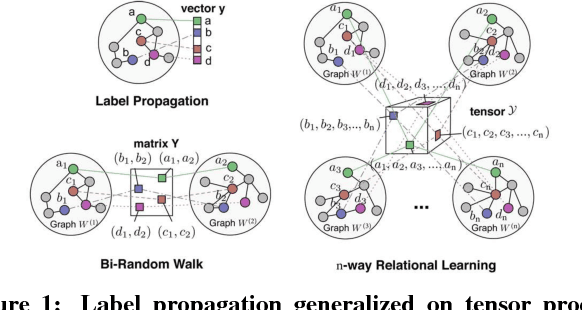
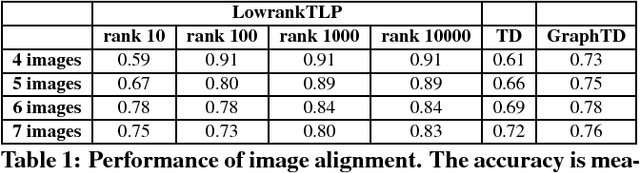
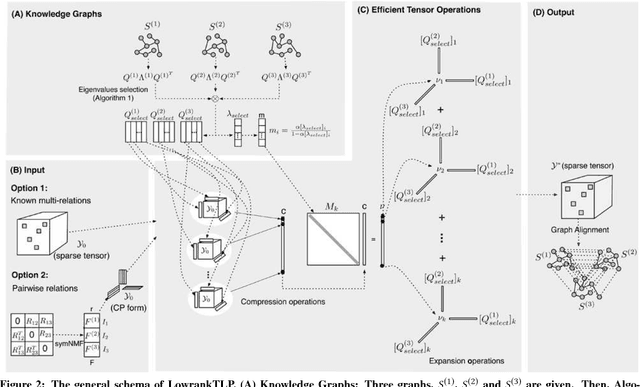
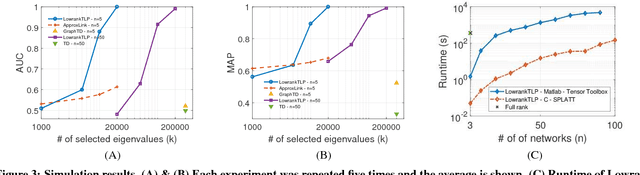
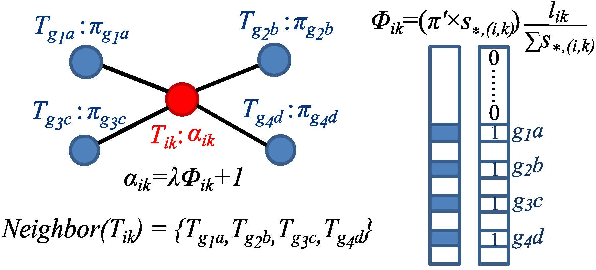
Label propagation on the tensor product of multiple graphs can infer multi-relations among the entities across the graphs by learning labels in a tensor. However, the tensor formulation is only empirically scalable up to three graphs due to the exponential complexity of computing tensors. In this paper, we propose an optimization formulation and a scalable Lowrank Tensor-based Label Propagation algorithm (LowrankTLP). The optimization formulation minimizes the rank-k approximation error for computing the closed-form solution of label propagation on a tensor product graph with efficient tensor computations used in LowrankTLP. LowrankTLP takes either a sparse tensor of known multi-relations or pairwise relations between each pair of graphs as the input to infer unknown multi-relations by semi-supervised learning on the tensor product graph. We also accelerate LowrankTLP with parallel tensor computation which enabled label propagation on a tensor product of 100 graphs of size 1000 within 150 seconds in simulation. LowrankTLP was also successfully applied to multi-relational learning for predicting author-paper-venue in publication records, alignment of several protein-protein interaction networks across species and alignment of segmented regions across up to 7 CT scan images. The experiments prove that LowrankTLP indeed well approximates the original label propagation with high scalability. Source code: https://github.com/kuanglab/LowrankTLP
Low-rank Label Propagation for Semi-supervised Learning with 100 Millions Samples
Feb 28, 2017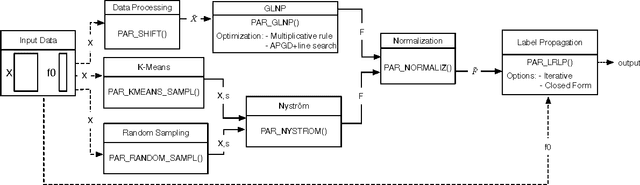

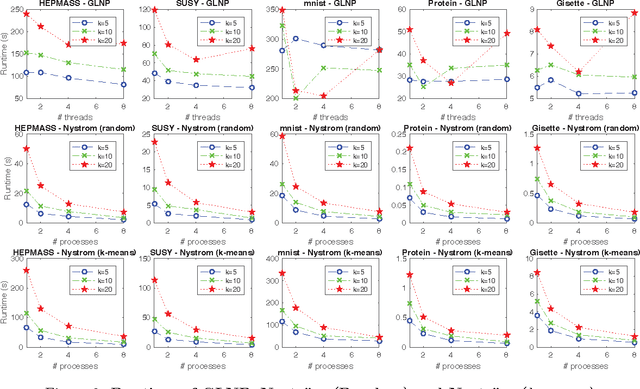
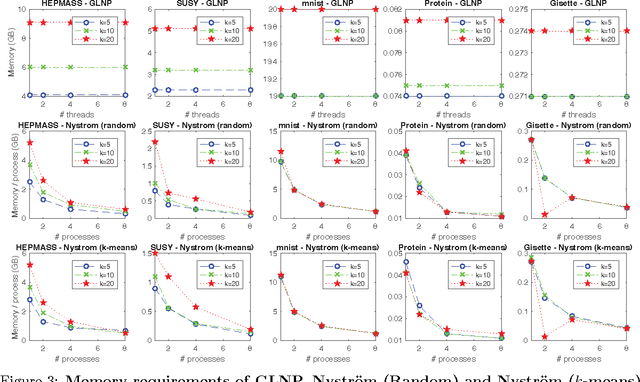
The success of semi-supervised learning crucially relies on the scalability to a huge amount of unlabelled data that are needed to capture the underlying manifold structure for better classification. Since computing the pairwise similarity between the training data is prohibitively expensive in most kinds of input data, currently, there is no general ready-to-use semi-supervised learning method/tool available for learning with tens of millions or more data points. In this paper, we adopted the idea of two low-rank label propagation algorithms, GLNP (Global Linear Neighborhood Propagation) and Kernel Nystr\"om Approximation, and implemented the parallelized version of the two algorithms accelerated with Nesterov's accelerated projected gradient descent for Big-data Label Propagation (BigLP). The parallel algorithms are tested on five real datasets ranging from 7000 to 10,000,000 in size and a simulation dataset of 100,000,000 samples. In the experiments, the implementation can scale up to datasets with 100,000,000 samples and hundreds of features and the algorithms also significantly improved the prediction accuracy when only a very small percentage of the data is labeled. The results demonstrate that the BigLP implementation is highly scalable to big data and effective in utilizing the unlabeled data for semi-supervised learning.
Network-based Isoform Quantification with RNA-Seq Data for Cancer Transcriptome Analysis
Sep 15, 2015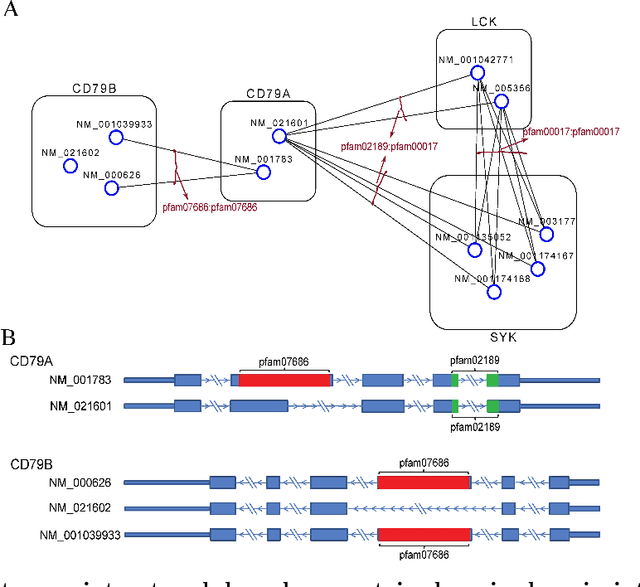


High-throughput mRNA sequencing (RNA-Seq) is widely used for transcript quantification of gene isoforms. Since RNA-Seq data alone is often not sufficient to accurately identify the read origins from the isoforms for quantification, we propose to explore protein domain-domain interactions as prior knowledge for integrative analysis with RNA-seq data. We introduce a Network-based method for RNA-Seq-based Transcript Quantification (Net-RSTQ) to integrate protein domain-domain interaction network with short read alignments for transcript abundance estimation. Based on our observation that the abundances of the neighboring isoforms by domain-domain interactions in the network are positively correlated, Net-RSTQ models the expression of the neighboring transcripts as Dirichlet priors on the likelihood of the observed read alignments against the transcripts in one gene. The transcript abundances of all the genes are then jointly estimated with alternating optimization of multiple EM problems. In simulation Net-RSTQ effectively improved isoform transcript quantifications when isoform co-expressions correlate with their interactions. qRT-PCR results on 25 multi-isoform genes in a stem cell line, an ovarian cancer cell line, and a breast cancer cell line also showed that Net-RSTQ estimated more consistent isoform proportions with RNA-Seq data. In the experiments on the RNA-Seq data in The Cancer Genome Atlas (TCGA), the transcript abundances estimated by Net-RSTQ are more informative for patient sample classification of ovarian cancer, breast cancer and lung cancer. All experimental results collectively support that Net-RSTQ is a promising approach for isoform quantification.
Inferring Disease and Gene Set Associations with Rank Coherence in Networks
Feb 18, 2011
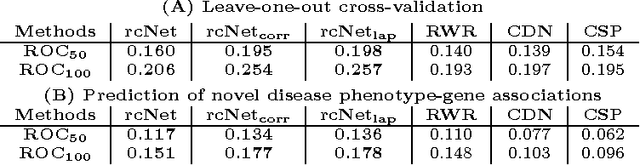

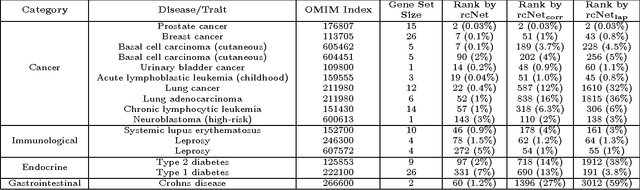
A computational challenge to validate the candidate disease genes identified in a high-throughput genomic study is to elucidate the associations between the set of candidate genes and disease phenotypes. The conventional gene set enrichment analysis often fails to reveal associations between disease phenotypes and the gene sets with a short list of poorly annotated genes, because the existing annotations of disease causative genes are incomplete. We propose a network-based computational approach called rcNet to discover the associations between gene sets and disease phenotypes. Assuming coherent associations between the genes ranked by their relevance to the query gene set, and the disease phenotypes ranked by their relevance to the hidden target disease phenotypes of the query gene set, we formulate a learning framework maximizing the rank coherence with respect to the known disease phenotype-gene associations. An efficient algorithm coupling ridge regression with label propagation, and two variants are introduced to find the optimal solution of the framework. We evaluated the rcNet algorithms and existing baseline methods with both leave-one-out cross-validation and a task of predicting recently discovered disease-gene associations in OMIM. The experiments demonstrated that the rcNet algorithms achieved the best overall rankings compared to the baselines. To further validate the reproducibility of the performance, we applied the algorithms to identify the target diseases of novel candidate disease genes obtained from recent studies of GWAS, DNA copy number variation analysis, and gene expression profiling. The algorithms ranked the target disease of the candidate genes at the top of the rank list in many cases across all the three case studies. The rcNet algorithms are available as a webtool for disease and gene set association analysis at http://compbio.cs.umn.edu/dgsa_rcNet.