 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeConvex Optimization for Binary Classifier Aggregation in Multiclass Problems
Jan 16, 2014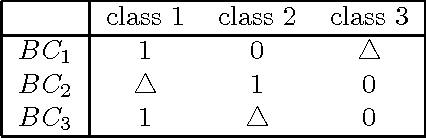
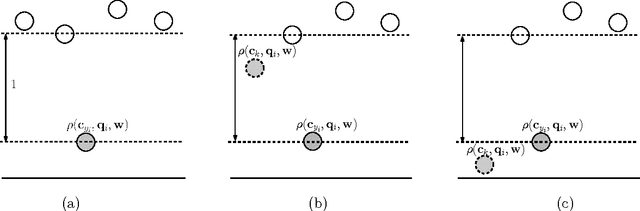
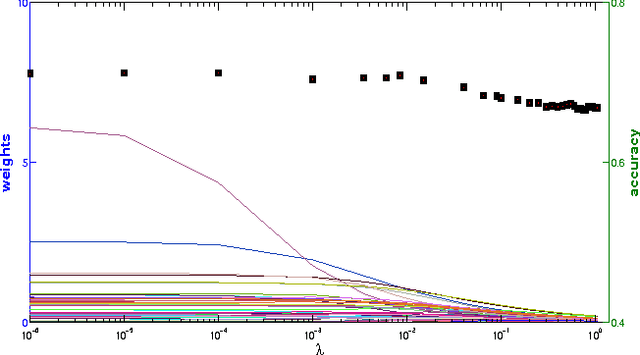
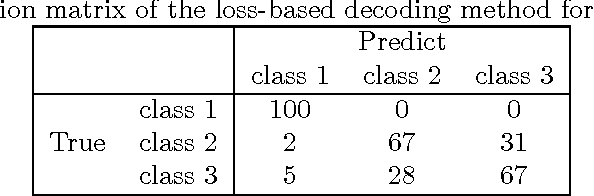
Multiclass problems are often decomposed into multiple binary problems that are solved by individual binary classifiers whose results are integrated into a final answer. Various methods, including all-pairs (APs), one-versus-all (OVA), and error correcting output code (ECOC), have been studied, to decompose multiclass problems into binary problems. However, little study has been made to optimally aggregate binary problems to determine a final answer to the multiclass problem. In this paper we present a convex optimization method for an optimal aggregation of binary classifiers to estimate class membership probabilities in multiclass problems. We model the class membership probability as a softmax function which takes a conic combination of discrepancies induced by individual binary classifiers, as an input. With this model, we formulate the regularized maximum likelihood estimation as a convex optimization problem, which is solved by the primal-dual interior point method. Connections of our method to large margin classifiers are presented, showing that the large margin formulation can be considered as a limiting case of our convex formulation. Numerical experiments on synthetic and real-world data sets demonstrate that our method outperforms existing aggregation methods as well as direct methods, in terms of the classification accuracy and the quality of class membership probability estimates.
Inferring Disease and Gene Set Associations with Rank Coherence in Networks
Feb 18, 2011
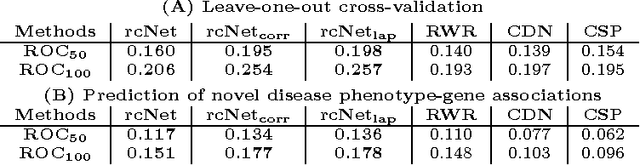

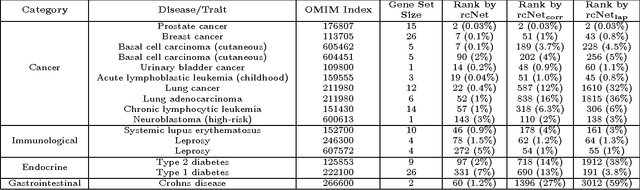
A computational challenge to validate the candidate disease genes identified in a high-throughput genomic study is to elucidate the associations between the set of candidate genes and disease phenotypes. The conventional gene set enrichment analysis often fails to reveal associations between disease phenotypes and the gene sets with a short list of poorly annotated genes, because the existing annotations of disease causative genes are incomplete. We propose a network-based computational approach called rcNet to discover the associations between gene sets and disease phenotypes. Assuming coherent associations between the genes ranked by their relevance to the query gene set, and the disease phenotypes ranked by their relevance to the hidden target disease phenotypes of the query gene set, we formulate a learning framework maximizing the rank coherence with respect to the known disease phenotype-gene associations. An efficient algorithm coupling ridge regression with label propagation, and two variants are introduced to find the optimal solution of the framework. We evaluated the rcNet algorithms and existing baseline methods with both leave-one-out cross-validation and a task of predicting recently discovered disease-gene associations in OMIM. The experiments demonstrated that the rcNet algorithms achieved the best overall rankings compared to the baselines. To further validate the reproducibility of the performance, we applied the algorithms to identify the target diseases of novel candidate disease genes obtained from recent studies of GWAS, DNA copy number variation analysis, and gene expression profiling. The algorithms ranked the target disease of the candidate genes at the top of the rank list in many cases across all the three case studies. The rcNet algorithms are available as a webtool for disease and gene set association analysis at http://compbio.cs.umn.edu/dgsa_rcNet.