 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeCross-Examination Framework: A Task-Agnostic Diagnostic for Information Fidelity in Text-to-Text Generation
Jan 27, 2026Traditional metrics like BLEU and BERTScore fail to capture semantic fidelity in generative text-to-text tasks. We adapt the Cross-Examination Framework (CEF) for a reference-free, multi-dimensional evaluation by treating the source and candidate as independent knowledge bases. CEF generates verifiable questions from each text and performs a cross-examination to derive three interpretable scores: Coverage, Conformity, and Consistency. Validated across translation, summarization and clinical note-generation, our framework identifies critical errors, such as content omissions and factual contradictions, missed by standard metrics. A key contribution is a systematic robustness analysis to select a stable judge model. Crucially, the strong correlation between our reference-free and with-reference modes validates CEF's reliability without gold references. Furthermore, human expert validation demonstrates that CEF mismatching questions align with meaning-altering semantic errors higher than with non-semantic errors, particularly excelling at identifying entity-based and relational distortions.
Overalignment in Frontier LLMs: An Empirical Study of Sycophantic Behaviour in Healthcare
Jan 26, 2026As LLMs are increasingly integrated into clinical workflows, their tendency for sycophancy, prioritizing user agreement over factual accuracy, poses significant risks to patient safety. While existing evaluations often rely on subjective datasets, we introduce a robust framework grounded in medical MCQA with verifiable ground truths. We propose the Adjusted Sycophancy Score, a novel metric that isolates alignment bias by accounting for stochastic model instability, or "confusability". Through an extensive scaling analysis of the Qwen-3 and Llama-3 families, we identify a clear scaling trajectory for resilience. Furthermore, we reveal a counter-intuitive vulnerability in reasoning-optimized "Thinking" models: while they demonstrate high vanilla accuracy, their internal reasoning traces frequently rationalize incorrect user suggestions under authoritative pressure. Our results across frontier models suggest that benchmark performance is not a proxy for clinical reliability, and that simplified reasoning structures may offer superior robustness against expert-driven sycophancy.
Do Instruction-Tuned Models Always Perform Better Than Base Models? Evidence from Math and Domain-Shifted Benchmarks
Jan 19, 2026Instruction finetuning is standard practice for improving LLM performance, yet it remains unclear whether it enhances reasoning or merely induces surface-level pattern matching. We investigate this by evaluating base and instruction-tuned models on standard math benchmarks, structurally perturbed variants, and domain-shifted tasks. Our analysis highlights two key (often overlooked) limitations of instruction tuning. First, the performance advantage is unstable and depends heavily on evaluation settings. In zero-shot CoT settings on GSM8K, base models consistently outperform instruction-tuned variants, with drops as high as 32.67\% (Llama3-70B). Instruction-tuned models only match or exceed this performance when provided with few-shot exemplars, suggesting a reliance on specific prompting patterns rather than intrinsic reasoning. Second, tuning gains are brittle under distribution shift. Our results show that base models surpass instruction-tuned variants on the domain-specific MedCalc benchmark. Additionally, instruction-tuned models show sharp declines on perturbed datasets, indicating sensitivity to prompt structure over robust reasoning.
Gene42: Long-Range Genomic Foundation Model With Dense Attention
Mar 20, 2025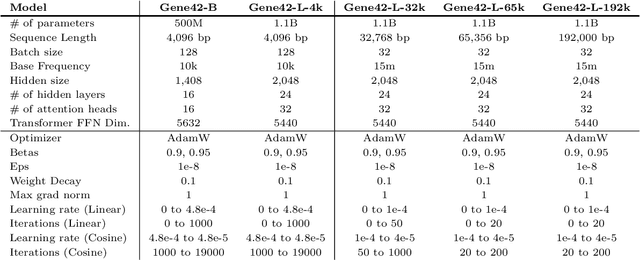
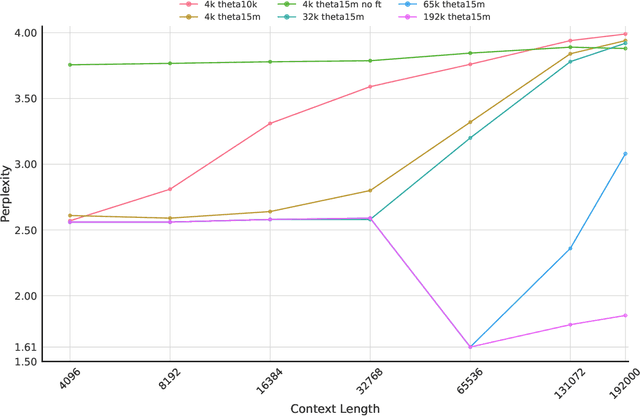
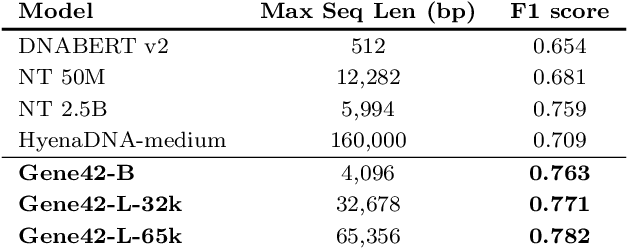
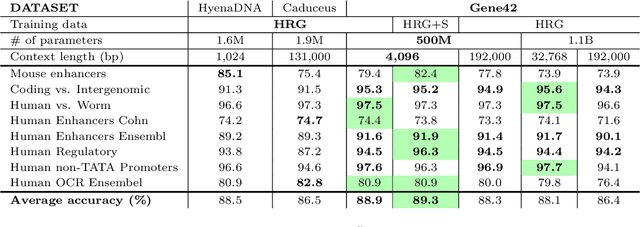
We introduce Gene42, a novel family of Genomic Foundation Models (GFMs) designed to manage context lengths of up to 192,000 base pairs (bp) at a single-nucleotide resolution. Gene42 models utilize a decoder-only (LLaMA-style) architecture with a dense self-attention mechanism. Initially trained on fixed-length sequences of 4,096 bp, our models underwent continuous pretraining to extend the context length to 192,000 bp. This iterative extension allowed for the comprehensive processing of large-scale genomic data and the capture of intricate patterns and dependencies within the human genome. Gene42 is the first dense attention model capable of handling such extensive long context lengths in genomics, challenging state-space models that often rely on convolutional operators among other mechanisms. Our pretrained models exhibit notably low perplexity values and high reconstruction accuracy, highlighting their strong ability to model genomic data. Extensive experiments on various genomic benchmarks have demonstrated state-of-the-art performance across multiple tasks, including biotype classification, regulatory region identification, chromatin profiling prediction, variant pathogenicity prediction, and species classification. The models are publicly available at huggingface.co/inceptionai.