 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAutomating tumor-infiltrating lymphocyte assessment in breast cancer histopathology images using QuPath: a transparent and accessible machine learning pipeline
Apr 23, 2025In this study, we built an end-to-end tumor-infiltrating lymphocytes (TILs) assessment pipeline within QuPath, demonstrating the potential of easily accessible tools to perform complex tasks in a fully automatic fashion. First, we trained a pixel classifier to segment tumor, tumor-associated stroma, and other tissue compartments in breast cancer H&E-stained whole-slide images (WSI) to isolate tumor-associated stroma for subsequent analysis. Next, we applied a pre-trained StarDist deep learning model in QuPath for cell detection and used the extracted cell features to train a binary classifier distinguishing TILs from other cells. To evaluate our TILs assessment pipeline, we calculated the TIL density in each WSI and categorized them as low, medium, or high TIL levels. Our pipeline was evaluated against pathologist-assigned TIL scores, achieving a Cohen's kappa of 0.71 on the external test set, corroborating previous research findings. These results confirm that existing software can offer a practical solution for the assessment of TILs in H&E-stained WSIs of breast cancer.
Open-source framework for detecting bias and overfitting for large pathology images
Mar 03, 2025Even foundational models that are trained on datasets with billions of data samples may develop shortcuts that lead to overfitting and bias. Shortcuts are non-relevant patterns in data, such as the background color or color intensity. So, to ensure the robustness of deep learning applications, there is a need for methods to detect and remove such shortcuts. Today's model debugging methods are time consuming since they often require customization to fit for a given model architecture in a specific domain. We propose a generalized, model-agnostic framework to debug deep learning models. We focus on the domain of histopathology, which has very large images that require large models - and therefore large computation resources. It can be run on a workstation with a commodity GPU. We demonstrate that our framework can replicate non-image shortcuts that have been found in previous work for self-supervised learning models, and we also identify possible shortcuts in a foundation model. Our easy to use tests contribute to the development of more reliable, accurate, and generalizable models for WSI analysis. Our framework is available as an open-source tool available on github.
A Lightweight and Extensible Cell Segmentation and Classification Model for Whole Slide Images
Feb 26, 2025
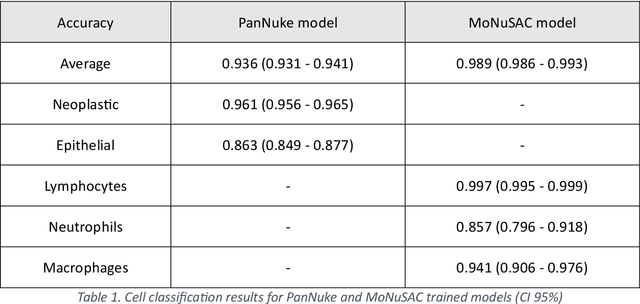

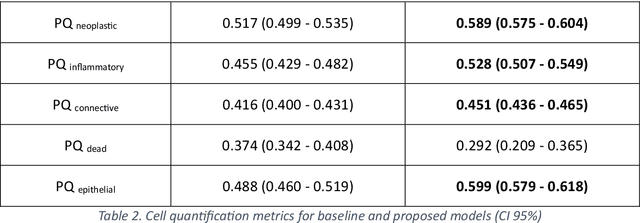
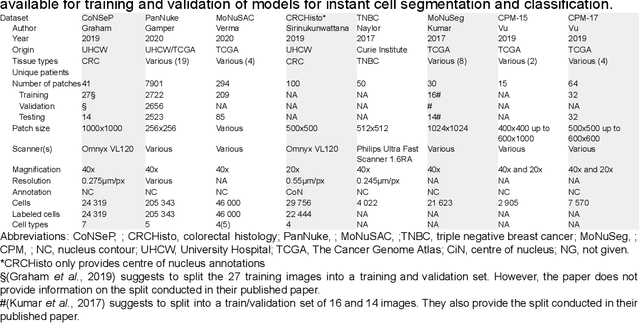
Developing clinically useful cell-level analysis tools in digital pathology remains challenging due to limitations in dataset granularity, inconsistent annotations, high computational demands, and difficulties integrating new technologies into workflows. To address these issues, we propose a solution that enhances data quality, model performance, and usability by creating a lightweight, extensible cell segmentation and classification model. First, we update data labels through cross-relabeling to refine annotations of PanNuke and MoNuSAC, producing a unified dataset with seven distinct cell types. Second, we leverage the H-Optimus foundation model as a fixed encoder to improve feature representation for simultaneous segmentation and classification tasks. Third, to address foundation models' computational demands, we distill knowledge to reduce model size and complexity while maintaining comparable performance. Finally, we integrate the distilled model into QuPath, a widely used open-source digital pathology platform. Results demonstrate improved segmentation and classification performance using the H-Optimus-based model compared to a CNN-based model. Specifically, average $R^2$ improved from 0.575 to 0.871, and average $PQ$ score improved from 0.450 to 0.492, indicating better alignment with actual cell counts and enhanced segmentation quality. The distilled model maintains comparable performance while reducing parameter count by a factor of 48. By reducing computational complexity and integrating into workflows, this approach may significantly impact diagnostics, reduce pathologist workload, and improve outcomes. Although the method shows promise, extensive validation is necessary prior to clinical deployment.
Fast TILs estimation in lung cancer WSIs based on semi-stochastic patch sampling
May 05, 2024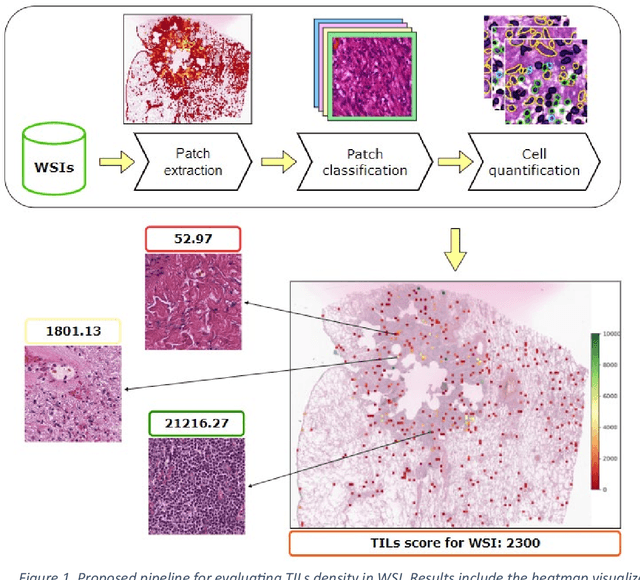
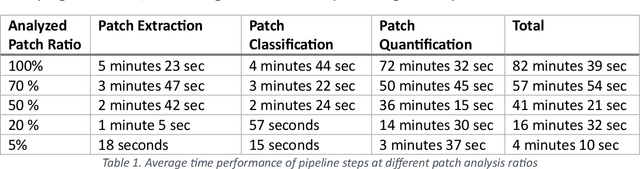
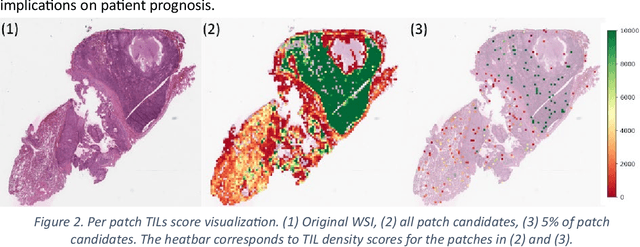
Addressing the critical need for accurate prognostic biomarkers in cancer treatment, quantifying tumor-infiltrating lymphocytes (TILs) in non-small cell lung cancer (NSCLC) presents considerable challenges. Manual TIL quantification in whole slide images (WSIs) is laborious and subject to variability, potentially undermining patient outcomes. Our study introduces an automated pipeline that utilizes semi-stochastic patch sampling, patch classification to retain prognostically relevant patches, and cell quantification using the HoVer-Net model to streamline the TIL evaluation process. This pipeline efficiently excludes approximately 70% of areas not relevant for prognosis and requires only 5% of the remaining patches to maintain prognostic accuracy (c-index 0.65 +- 0.01). The computational efficiency achieved does not sacrifice prognostic accuracy, as demonstrated by the TILs score's strong correlation with patient survival, which surpasses traditional CD8 IHC scoring methods. While the pipeline demonstrates potential for enhancing NSCLC prognostication and personalization of treatment, comprehensive clinical validation is still required. Future research should focus on verifying its broader clinical utility and investigating additional biomarkers to improve NSCLC prognosis.
Publicly available datasets of breast histopathology H&E whole-slide images: A systematic review
Jun 02, 2023Advancements in digital pathology and computing resources have made a significant impact in the field of computational pathology for breast cancer diagnosis and treatment. However, access to high-quality labeled histopathological images of breast cancer is a big challenge that limits the development of accurate and robust deep learning models. In this systematic review, we identified the publicly available datasets of breast H&E stained whole-slide images (WSI) that can be used to develop deep learning algorithms. We systematically searched nine scientific literature databases and nine research data repositories. We found twelve publicly available datasets, containing 5153 H&E WSIs of breast cancer. Moreover, we reported image metadata and characteristics for each dataset to assist researchers in selecting proper datasets for specific tasks in breast cancer computational pathology. In addition, we compiled a list of patch and private datasets that were used in the included articles as a supplementary resource for researchers. Notably, 22% of the included articles utilized multiple datasets, and only 12% of the articles used an external validation set, suggesting that the performance of other developed models may be susceptible to overestimation. The TCGA-BRCA was used in 47.4% of the selected studies. This dataset has a considerable selection bias that can impact the robustness and generalizability of the trained algorithms. There is also a lack of consistent metadata reporting of breast WSI datasets that can be an issue in developing accurate deep learning models, indicating the necessity of establishing explicit guidelines for documenting breast WSI dataset characteristics and metadata.
A Pragmatic Machine Learning Approach to Quantify Tumor Infiltrating Lymphocytes in Whole Slide Images
Feb 14, 2022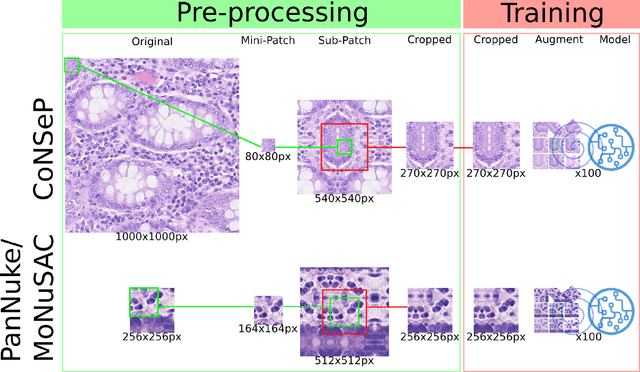
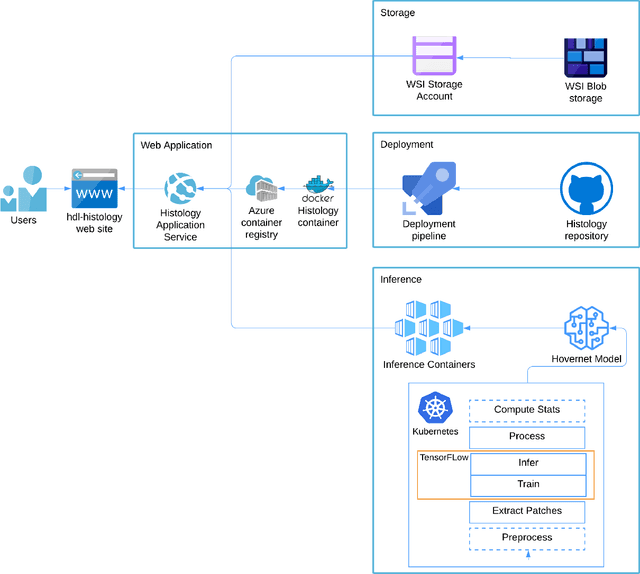
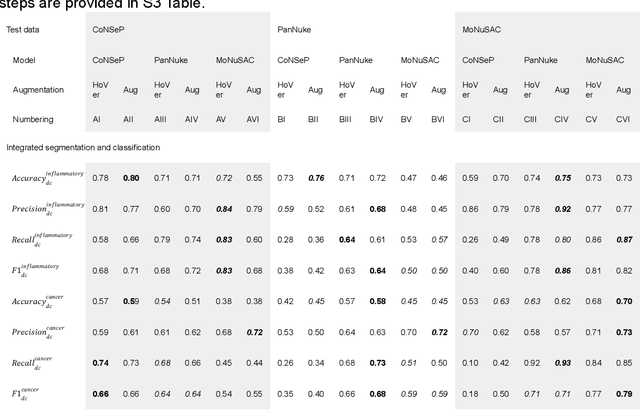
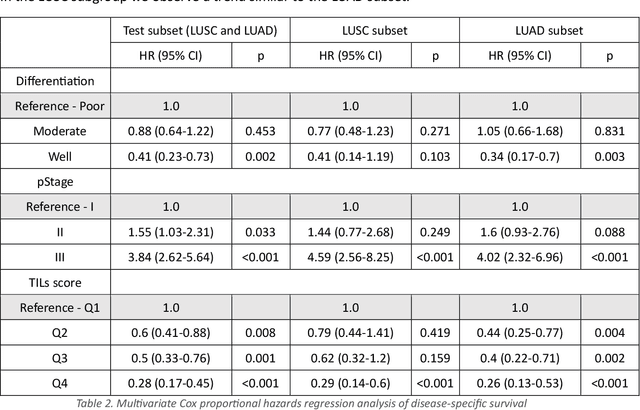
Increased levels of tumor infiltrating lymphocytes (TILs) in cancer tissue indicate favourable outcomes in many types of cancer. Manual quantification of immune cells is inaccurate and time consuming for pathologists. Our aim is to leverage a computational solution to automatically quantify TILs in whole slide images (WSIs) of standard diagnostic haematoxylin and eosin stained sections (H&E slides) from lung cancer patients. Our approach is to transfer an open source machine learning method for segmentation and classification of nuclei in H&E slides trained on public data to TIL quantification without manual labeling of our data. Our results show that additional augmentation improves model transferability when training on few samples/limited tissue types. Models trained with sufficient samples/tissue types do not benefit from our additional augmentation policy. Further, the resulting TIL quantification correlates to patient prognosis and compares favorably to the current state-of-the-art method for immune cell detection in non-small lung cancer (current standard CD8 cells in DAB stained TMAs HR 0.34 95% CI 0.17-0.68 vs TILs in HE WSIs: HoVer-Net PanNuke Aug Model HR 0.30 95% CI 0.15-0.60, HoVer-Net MoNuSAC Aug model HR 0.27 95% CI 0.14-0.53). Moreover, we implemented a cloud based system to train, deploy and visually inspect machine learning based annotation for H&E slides. Our pragmatic approach bridges the gap between machine learning research, translational clinical research and clinical implementation. However, validation in prospective studies is needed to assert that the method works in a clinical setting.
Occode: an end-to-end machine learning pipeline for transcription of historical population censuses
Jun 07, 2021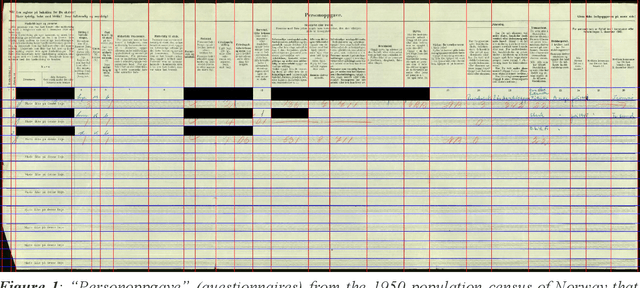
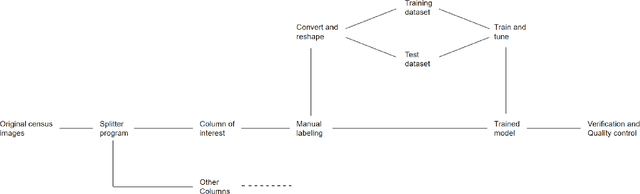
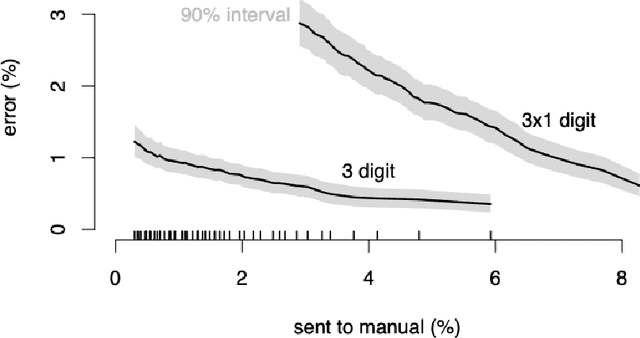
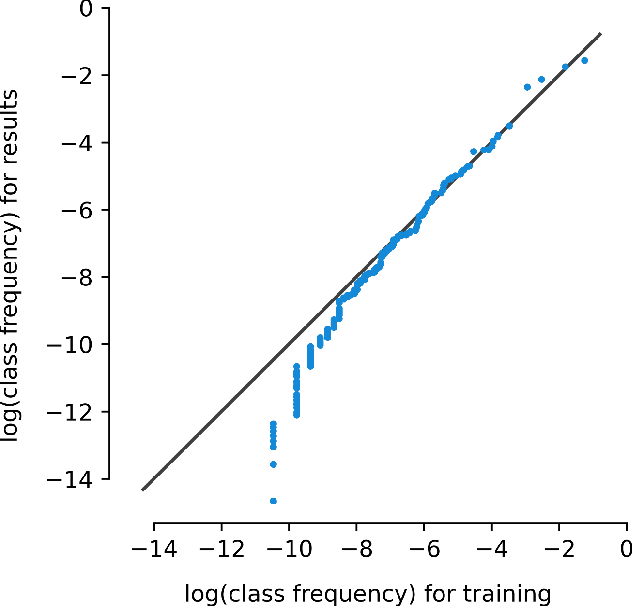
Machine learning approaches achieve high accuracy for text recognition and are therefore increasingly used for the transcription of handwritten historical sources. However, using machine learning in production requires a streamlined end-to-end machine learning pipeline that scales to the dataset size, and a model that achieves high accuracy with few manual transcriptions. In addition, the correctness of the model results must be verified. This paper describes our lessons learned developing, tuning, and using the Occode end-to-end machine learning pipeline for transcribing 7,3 million rows with handwritten occupation codes in the Norwegian 1950 population census. We achieve an accuracy of 97% for the automatically transcribed codes, and we send 3% of the codes for manual verification. We verify that the occupation code distribution found in our result matches the distribution found in our training data which should be representative for the census as a whole. We believe our approach and lessons learned are useful for other transcription projects that plan to use machine learning in production. The source code is available at: https://github.com/uit-hdl/rhd-codes