 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeDeeptime: a Python library for machine learning dynamical models from time series data
Oct 28, 2021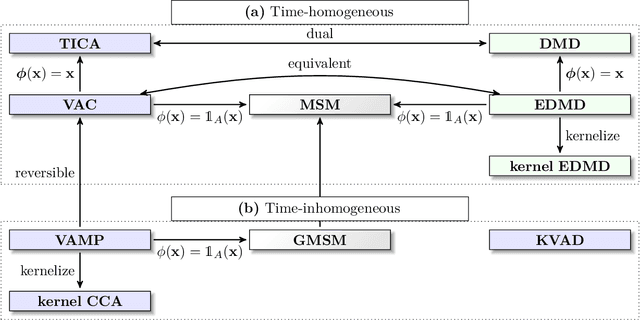

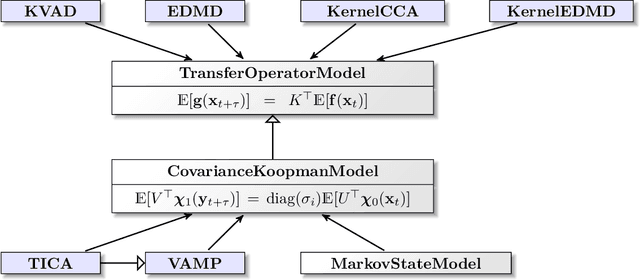
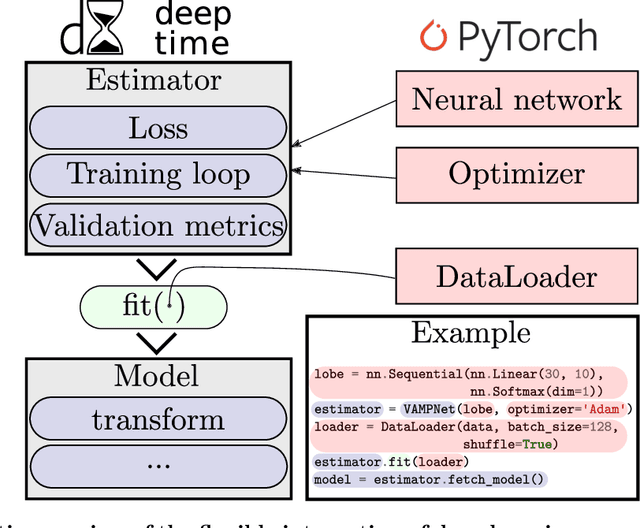
Generation and analysis of time-series data is relevant to many quantitative fields ranging from economics to fluid mechanics. In the physical sciences, structures such as metastable and coherent sets, slow relaxation processes, collective variables dominant transition pathways or manifolds and channels of probability flow can be of great importance for understanding and characterizing the kinetic, thermodynamic and mechanistic properties of the system. Deeptime is a general purpose Python library offering various tools to estimate dynamical models based on time-series data including conventional linear learning methods, such as Markov state models (MSMs), Hidden Markov Models and Koopman models, as well as kernel and deep learning approaches such as VAMPnets and deep MSMs. The library is largely compatible with scikit-learn, having a range of Estimator classes for these different models, but in contrast to scikit-learn also provides deep Model classes, e.g. in the case of an MSM, which provide a multitude of analysis methods to compute interesting thermodynamic, kinetic and dynamical quantities, such as free energies, relaxation times and transition paths. The library is designed for ease of use but also easily maintainable and extensible code. In this paper we introduce the main features and structure of the deeptime software.
Generating valid Euclidean distance matrices
Nov 15, 2019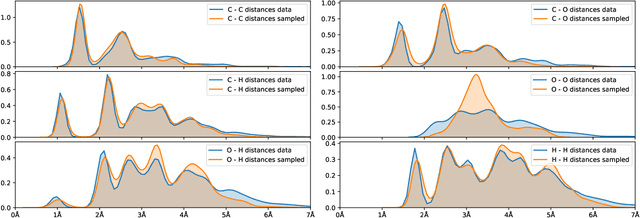
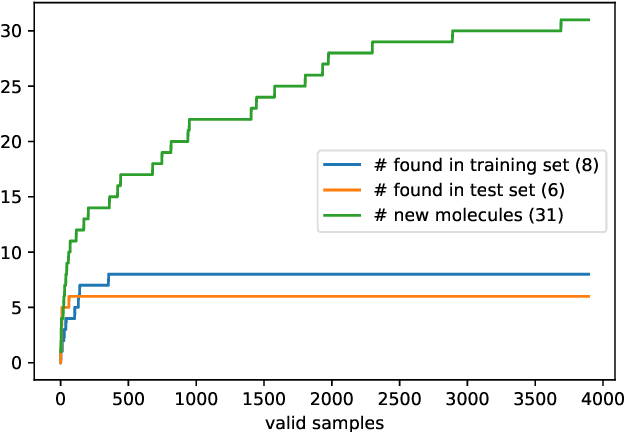
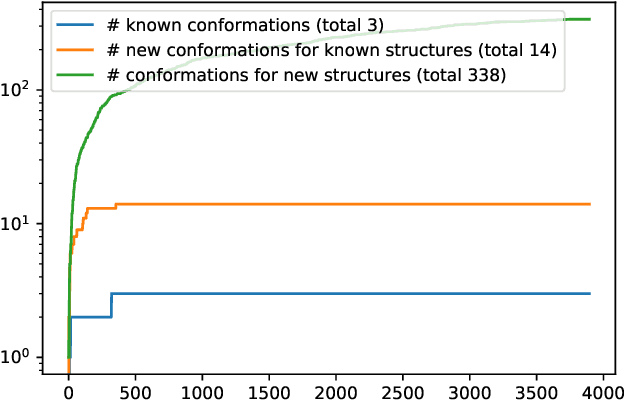
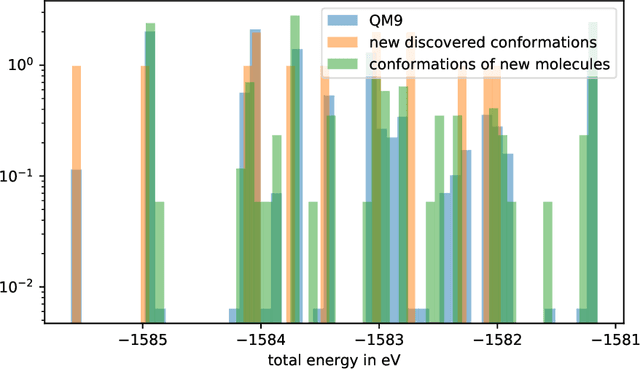
Generating point clouds, e.g., molecular structures, in arbitrary rotations, translations, and enumerations remains a challenging task. Meanwhile, neural networks utilizing symmetry invariant layers have been shown to be able to optimize their training objective in a data-efficient way. In this spirit, we present an architecture which allows to produce valid Euclidean distance matrices, which by construction are already invariant under rotation and translation of the described object. Motivated by the goal to generate molecular structures in Cartesian space, we use this architecture to construct a Wasserstein GAN utilizing a permutation invariant critic network. This makes it possible to generate molecular structures in a one-shot fashion by producing Euclidean distance matrices which have a three-dimensional embedding.
Variational Selection of Features for Molecular Kinetics
Nov 28, 2018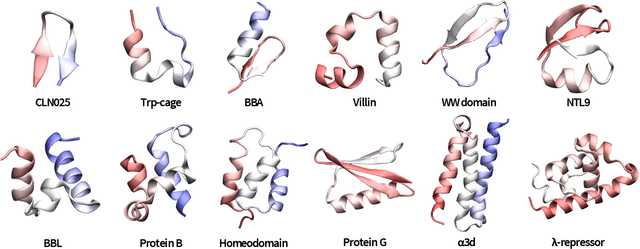

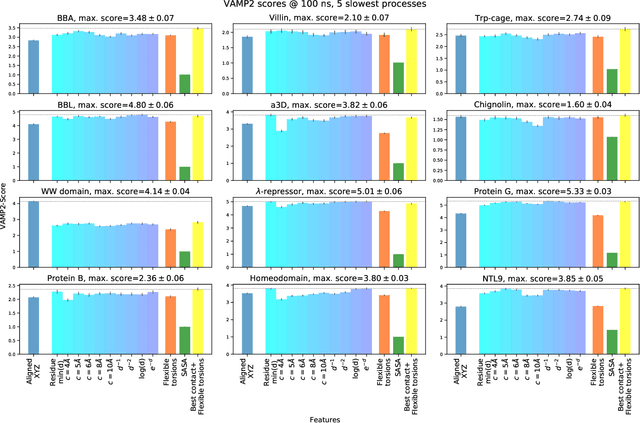
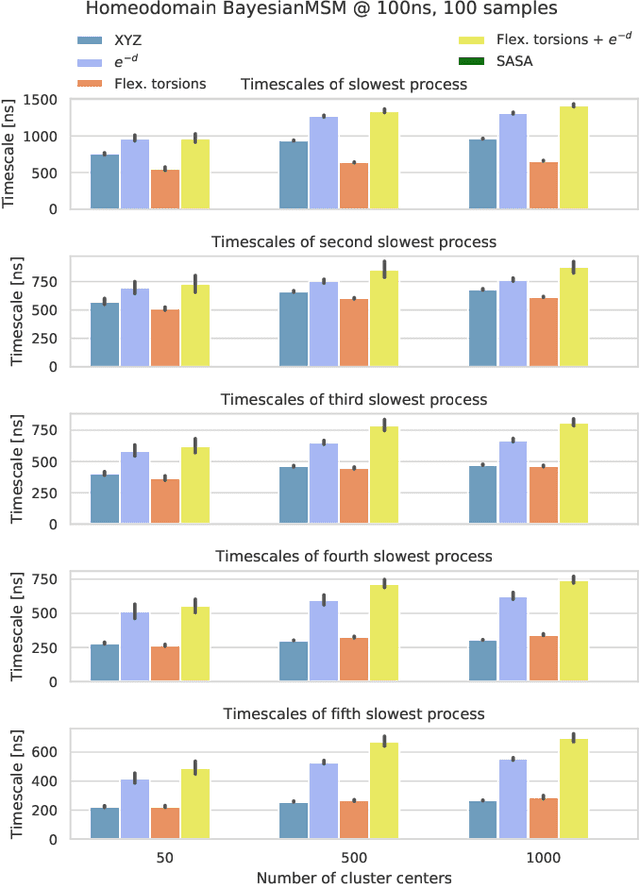
The modeling of atomistic biomolecular simulations using kinetic models such as Markov state models (MSMs) has had many notable algorithmic advances in recent years. The variational principle has opened the door for a nearly fully automated toolkit for selecting models that predict the long-time kinetics from molecular dynamics simulations. However, one yet-unoptimized step of the pipeline involves choosing the features, or collective variables, from which the model should be constructed. In order to build intuitive models, these collective variables are often sought to be interpretable and familiar features, such as torsional angles or contact distances in a protein structure. However, previous approaches for evaluating the chosen features rely on constructing a full MSM, which in turn requires additional hyperparameters to be chosen, and hence leads to a computationally expensive framework. Here, we present a method to optimize the feature choice directly, without requiring the construction of the final kinetic model. We demonstrate our rigorous preprocessing algorithm on a canonical set of twelve fast-folding protein simulations, and show that our procedure leads to more efficient model selection.