 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeVariational Selection of Features for Molecular Kinetics
Nov 28, 2018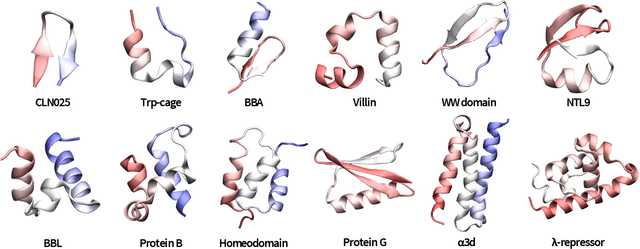

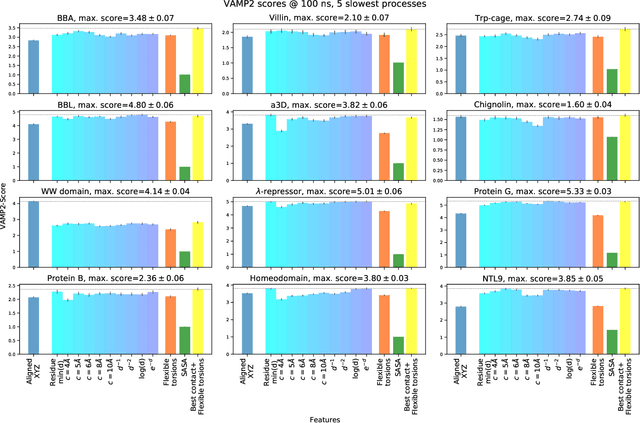
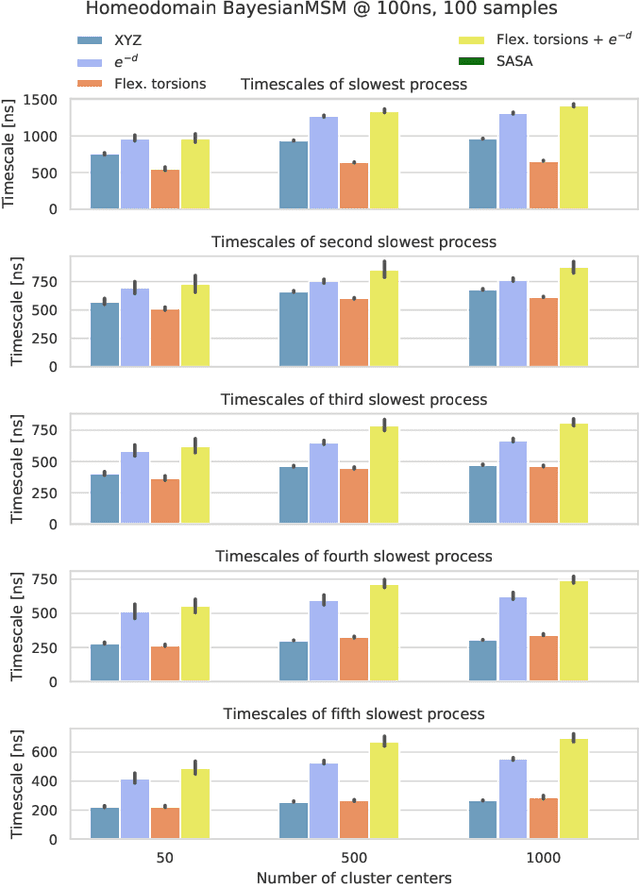
The modeling of atomistic biomolecular simulations using kinetic models such as Markov state models (MSMs) has had many notable algorithmic advances in recent years. The variational principle has opened the door for a nearly fully automated toolkit for selecting models that predict the long-time kinetics from molecular dynamics simulations. However, one yet-unoptimized step of the pipeline involves choosing the features, or collective variables, from which the model should be constructed. In order to build intuitive models, these collective variables are often sought to be interpretable and familiar features, such as torsional angles or contact distances in a protein structure. However, previous approaches for evaluating the chosen features rely on constructing a full MSM, which in turn requires additional hyperparameters to be chosen, and hence leads to a computationally expensive framework. Here, we present a method to optimize the feature choice directly, without requiring the construction of the final kinetic model. We demonstrate our rigorous preprocessing algorithm on a canonical set of twelve fast-folding protein simulations, and show that our procedure leads to more efficient model selection.