 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeFASTR: Reimagining FASTQ via Compact Image-inspired Representation
Jan 23, 2026Motivation: High-throughput sequencing (HTS) enables population-scale genomics but generates massive datasets, creating bottlenecks in storage, transfer, and analysis. FASTQ, the standard format for over two decades, stores one byte per base and one byte per quality score, leading to inefficient I/O, high storage costs, and redundancy. Existing compression tools can mitigate some issues, but often introduce costly decompression or complex dependency issues. Results: We introduce FASTR, a lossless, computation-native successor to FASTQ that encodes each nucleotide together with its base quality score into a single 8-bit value. FASTR reduces file size by at least 2x while remaining fully reversible and directly usable for downstream analyses. Applying general-purpose compression tools on FASTR consistently yields higher compression ratios, 2.47, 3.64, and 4.8x faster compression, and 2.34, 1.96, 1.75x faster decompression than on FASTQ across Illumina, HiFi, and ONT reads. FASTR is machine-learning-ready, allowing reads to be consumed directly as numerical vectors or image-like representations. We provide a highly parallel software ecosystem for FASTQ-FASTR conversion and show that FASTR integrates with existing tools, such as minimap2, with minimal interface changes and no performance overhead. By eliminating decompression costs and reducing data movement, FASTR lays the foundation for scalable genomics analyses and real-time sequencing workflows. Availability and Implementation: https://github.com/ALSER-Lab/FASTR
TargetCall: Eliminating the Wasted Computation in Basecalling via Pre-Basecalling Filtering
Dec 09, 2022Basecalling is an essential step in nanopore sequencing analysis where the raw signals of nanopore sequencers are converted into nucleotide sequences, i.e., reads. State-of-the-art basecallers employ complex deep learning models to achieve high basecalling accuracy. This makes basecalling computationally-inefficient and memory-hungry; bottlenecking the entire genome analysis pipeline. However, for many applications, the majority of reads do no match the reference genome of interest (i.e., target reference) and thus are discarded in later steps in the genomics pipeline, wasting the basecalling computation. To overcome this issue, we propose TargetCall, the first fast and widely-applicable pre-basecalling filter to eliminate the wasted computation in basecalling. TargetCall's key idea is to discard reads that will not match the target reference (i.e., off-target reads) prior to basecalling. TargetCall consists of two main components: (1) LightCall, a lightweight neural network basecaller that produces noisy reads; and (2) Similarity Check, which labels each of these noisy reads as on-target or off-target by matching them to the target reference. TargetCall filters out all off-target reads before basecalling; and the highly-accurate but slow basecalling is performed only on the raw signals whose noisy reads are labeled as on-target. Our thorough experimental evaluations using both real and simulated data show that TargetCall 1) improves the end-to-end basecalling performance of the state-of-the-art basecaller by 3.31x while maintaining high (98.88%) sensitivity in keeping on-target reads, 2) maintains high accuracy in downstream analysis, 3) precisely filters out up to 94.71% of off-target reads, and 4) achieves better performance, sensitivity, and generality compared to prior works. We freely open-source TargetCall at https://github.com/CMU-SAFARI/TargetCall.
ApHMM: Accelerating Profile Hidden Markov Models for Fast and Energy-Efficient Genome Analysis
Jul 20, 2022
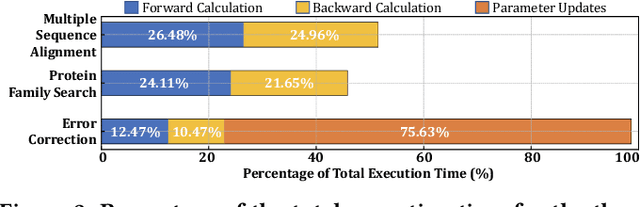
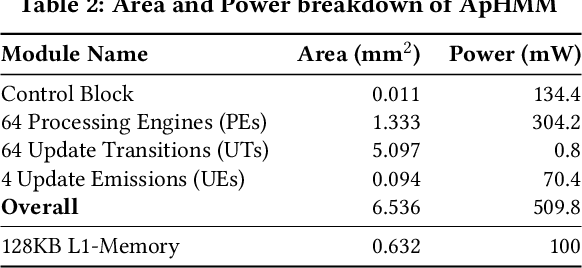
Profile hidden Markov models (pHMMs) are widely used in many bioinformatics applications to accurately identify similarities between biological sequences (e.g., DNA or protein sequences). PHMMs use a commonly-adopted and highly-accurate method, called the Baum-Welch algorithm, to calculate these similarities. However, the Baum-Welch algorithm is computationally expensive, and existing works provide either software- or hardware-only solutions for a fixed pHMM design. When we analyze the state-of-the-art works, we find that there is a pressing need for a flexible, high-performant, and energy-efficient hardware-software co-design to efficiently and effectively solve all the major inefficiencies in the Baum-Welch algorithm for pHMMs. We propose ApHMM, the first flexible acceleration framework that can significantly reduce computational and energy overheads of the Baum-Welch algorithm for pHMMs. ApHMM leverages hardware-software co-design to solve the major inefficiencies in the Baum-Welch algorithm by 1) designing a flexible hardware to support different pHMMs designs, 2) exploiting the predictable data dependency pattern in an on-chip memory with memoization techniques, 3) quickly eliminating negligible computations with a hardware-based filter, and 4) minimizing the redundant computations. We implement our 1) hardware-software optimizations on a specialized hardware and 2) software optimizations for GPUs to provide the first flexible Baum-Welch accelerator for pHMMs. ApHMM provides significant speedups of 15.55x-260.03x, 1.83x-5.34x, and 27.97x compared to CPU, GPU, and FPGA implementations of the Baum-Welch algorithm, respectively. ApHMM outperforms the state-of-the-art CPU implementations of three important bioinformatics applications, 1) error correction, 2) protein family search, and 3) multiple sequence alignment, by 1.29x-59.94x, 1.03x-1.75x, and 1.03x-1.95x, respectively.
COVIDHunter: COVID-19 pandemic wave prediction and mitigation via seasonality-aware modeling
Jun 14, 2022



Early detection and isolation of COVID-19 patients are essential for successful implementation of mitigation strategies and eventually curbing the disease spread. With a limited number of daily COVID-19 tests performed in every country, simulating the COVID-19 spread along with the potential effect of each mitigation strategy currently remains one of the most effective ways in managing the healthcare system and guiding policy-makers. We introduce COVIDHunter, a flexible and accurate COVID-19 outbreak simulation model that evaluates the current mitigation measures that are applied to a region, predicts COVID-19 statistics (the daily number of cases, hospitalizations, and deaths), and provides suggestions on what strength the upcoming mitigation measure should be. The key idea of COVIDHunter is to quantify the spread of COVID-19 in a geographical region by simulating the average number of new infections caused by an infected person considering the effect of external factors, such as environmental conditions (e.g., climate, temperature, humidity), different variants of concern, vaccination rate, and mitigation measures. Using Switzerland as a case study, COVIDHunter estimates that we are experiencing a deadly new wave that will peak on 26 January 2022, which is very similar in numbers to the wave we had in February 2020. The policy-makers have only one choice that is to increase the strength of the currently applied mitigation measures for 30 days. Unlike existing models, the COVIDHunter model accurately monitors and predicts the daily number of cases, hospitalizations, and deaths due to COVID-19. Our model is flexible to configure and simple to modify for modeling different scenarios under different environmental conditions and mitigation measures. We release the source code of the COVIDHunter implementation at https://github.com/CMU-SAFARI/COVIDHunter.
GateKeeper-GPU: Fast and Accurate Pre-Alignment Filtering in Short Read Mapping
Mar 31, 2021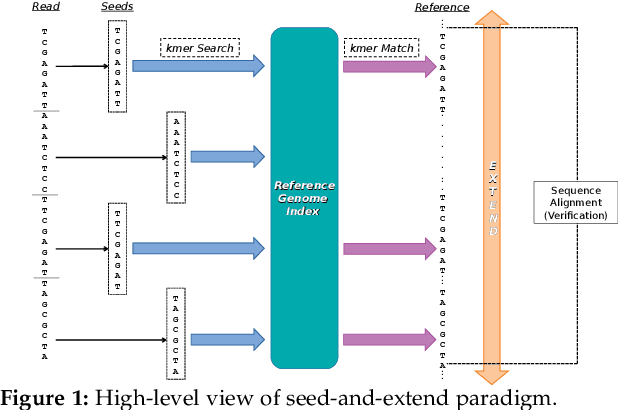
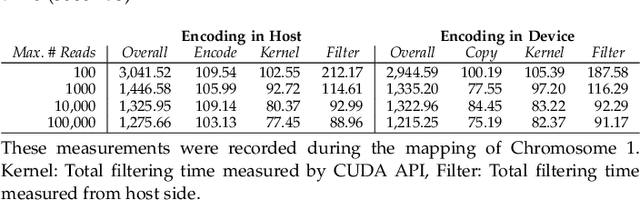

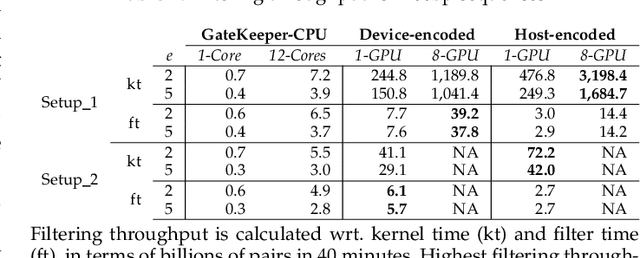
At the last step of short read mapping, the candidate locations of the reads on the reference genome are verified to compute their differences from the corresponding reference segments using sequence alignment algorithms. Calculating the similarities and differences between two sequences is still computationally expensive since approximate string matching techniques traditionally inherit dynamic programming algorithms with quadratic time and space complexity. We introduce GateKeeper-GPU, a fast and accurate pre-alignment filter that efficiently reduces the need for expensive sequence alignment. GateKeeper-GPU provides two main contributions: first, improving the filtering accuracy of GateKeeper(state-of-the-art lightweight pre-alignment filter), second, exploiting the massive parallelism provided by the large number of GPU threads of modern GPUs to examine numerous sequence pairs rapidly and concurrently. GateKeeper-GPU accelerates the sequence alignment by up to 2.9x and provides up to 1.4x speedup to the end-to-end execution time of a comprehensive read mapper (mrFAST). GateKeeper-GPU is available at https://github.com/BilkentCompGen/GateKeeper-GPU
COVIDHunter: An Accurate, Flexible, and Environment-Aware Open-Source COVID-19 Outbreak Simulation Model
Feb 06, 2021Motivation: Early detection and isolation of COVID-19 patients are essential for successful implementation of mitigation strategies and eventually curbing the disease spread. With a limited number of daily COVID19 tests performed in every country, simulating the COVID-19 spread along with the potential effect of each mitigation strategy currently remains one of the most effective ways in managing the healthcare system and guiding policy-makers. We introduce COVIDHunter, a flexible and accurate COVID-19 outbreak simulation model that evaluates the current mitigation measures that are applied to a region and provides suggestions on what strength the upcoming mitigation measure should be. The key idea of COVIDHunter is to quantify the spread of COVID-19 in a geographical region by simulating the average number of new infections caused by an infected person considering the effect of external factors, such as environmental conditions (e.g., climate, temperature, humidity) and mitigation measures. Results: Using Switzerland as a case study, COVIDHunter estimates that the policy-makers need to keep the current mitigation measures for at least 30 days to prevent demand from quickly exceeding existing hospital capacity. Relaxing the mitigation measures by 50% for 30 days increases both the daily capacity need for hospital beds and daily number of deaths exponentially by an average of 23.8x, who may occupy ICU beds and ventilators for a period of time. Unlike existing models, the COVIDHunter model accurately monitors and predicts the daily number of cases, hospitalizations, and deaths due to COVID-19. Our model is flexible to configure and simple to modify for modeling different scenarios under different environmental conditions and mitigation measures. Availability: https://github.com/CMU-SAFARI/COVIDHunter