 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeApHMM: Accelerating Profile Hidden Markov Models for Fast and Energy-Efficient Genome Analysis
Paper and Code

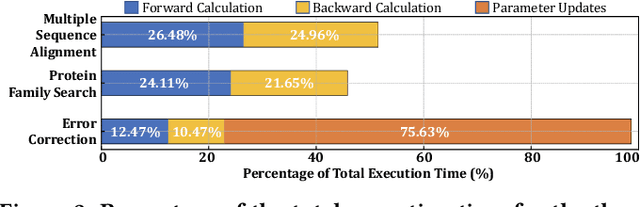
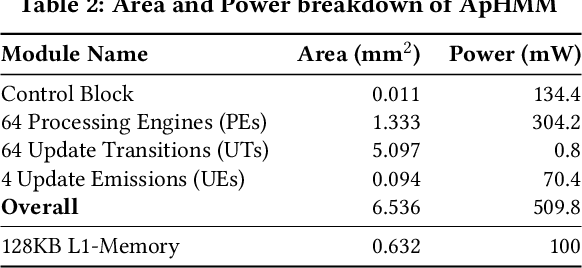
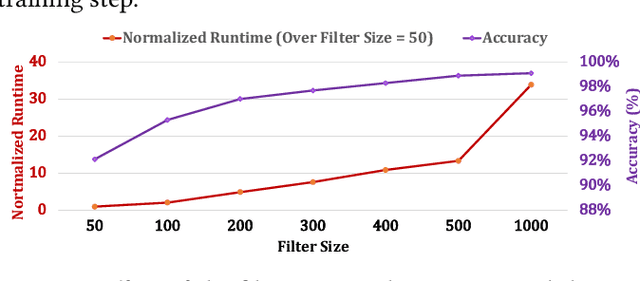
Profile hidden Markov models (pHMMs) are widely used in many bioinformatics applications to accurately identify similarities between biological sequences (e.g., DNA or protein sequences). PHMMs use a commonly-adopted and highly-accurate method, called the Baum-Welch algorithm, to calculate these similarities. However, the Baum-Welch algorithm is computationally expensive, and existing works provide either software- or hardware-only solutions for a fixed pHMM design. When we analyze the state-of-the-art works, we find that there is a pressing need for a flexible, high-performant, and energy-efficient hardware-software co-design to efficiently and effectively solve all the major inefficiencies in the Baum-Welch algorithm for pHMMs. We propose ApHMM, the first flexible acceleration framework that can significantly reduce computational and energy overheads of the Baum-Welch algorithm for pHMMs. ApHMM leverages hardware-software co-design to solve the major inefficiencies in the Baum-Welch algorithm by 1) designing a flexible hardware to support different pHMMs designs, 2) exploiting the predictable data dependency pattern in an on-chip memory with memoization techniques, 3) quickly eliminating negligible computations with a hardware-based filter, and 4) minimizing the redundant computations. We implement our 1) hardware-software optimizations on a specialized hardware and 2) software optimizations for GPUs to provide the first flexible Baum-Welch accelerator for pHMMs. ApHMM provides significant speedups of 15.55x-260.03x, 1.83x-5.34x, and 27.97x compared to CPU, GPU, and FPGA implementations of the Baum-Welch algorithm, respectively. ApHMM outperforms the state-of-the-art CPU implementations of three important bioinformatics applications, 1) error correction, 2) protein family search, and 3) multiple sequence alignment, by 1.29x-59.94x, 1.03x-1.75x, and 1.03x-1.95x, respectively.