 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeFASTR: Reimagining FASTQ via Compact Image-inspired Representation
Jan 23, 2026Motivation: High-throughput sequencing (HTS) enables population-scale genomics but generates massive datasets, creating bottlenecks in storage, transfer, and analysis. FASTQ, the standard format for over two decades, stores one byte per base and one byte per quality score, leading to inefficient I/O, high storage costs, and redundancy. Existing compression tools can mitigate some issues, but often introduce costly decompression or complex dependency issues. Results: We introduce FASTR, a lossless, computation-native successor to FASTQ that encodes each nucleotide together with its base quality score into a single 8-bit value. FASTR reduces file size by at least 2x while remaining fully reversible and directly usable for downstream analyses. Applying general-purpose compression tools on FASTR consistently yields higher compression ratios, 2.47, 3.64, and 4.8x faster compression, and 2.34, 1.96, 1.75x faster decompression than on FASTQ across Illumina, HiFi, and ONT reads. FASTR is machine-learning-ready, allowing reads to be consumed directly as numerical vectors or image-like representations. We provide a highly parallel software ecosystem for FASTQ-FASTR conversion and show that FASTR integrates with existing tools, such as minimap2, with minimal interface changes and no performance overhead. By eliminating decompression costs and reducing data movement, FASTR lays the foundation for scalable genomics analyses and real-time sequencing workflows. Availability and Implementation: https://github.com/ALSER-Lab/FASTR
TargetCall: Eliminating the Wasted Computation in Basecalling via Pre-Basecalling Filtering
Dec 09, 2022Basecalling is an essential step in nanopore sequencing analysis where the raw signals of nanopore sequencers are converted into nucleotide sequences, i.e., reads. State-of-the-art basecallers employ complex deep learning models to achieve high basecalling accuracy. This makes basecalling computationally-inefficient and memory-hungry; bottlenecking the entire genome analysis pipeline. However, for many applications, the majority of reads do no match the reference genome of interest (i.e., target reference) and thus are discarded in later steps in the genomics pipeline, wasting the basecalling computation. To overcome this issue, we propose TargetCall, the first fast and widely-applicable pre-basecalling filter to eliminate the wasted computation in basecalling. TargetCall's key idea is to discard reads that will not match the target reference (i.e., off-target reads) prior to basecalling. TargetCall consists of two main components: (1) LightCall, a lightweight neural network basecaller that produces noisy reads; and (2) Similarity Check, which labels each of these noisy reads as on-target or off-target by matching them to the target reference. TargetCall filters out all off-target reads before basecalling; and the highly-accurate but slow basecalling is performed only on the raw signals whose noisy reads are labeled as on-target. Our thorough experimental evaluations using both real and simulated data show that TargetCall 1) improves the end-to-end basecalling performance of the state-of-the-art basecaller by 3.31x while maintaining high (98.88%) sensitivity in keeping on-target reads, 2) maintains high accuracy in downstream analysis, 3) precisely filters out up to 94.71% of off-target reads, and 4) achieves better performance, sensitivity, and generality compared to prior works. We freely open-source TargetCall at https://github.com/CMU-SAFARI/TargetCall.
GateKeeper-GPU: Fast and Accurate Pre-Alignment Filtering in Short Read Mapping
Mar 31, 2021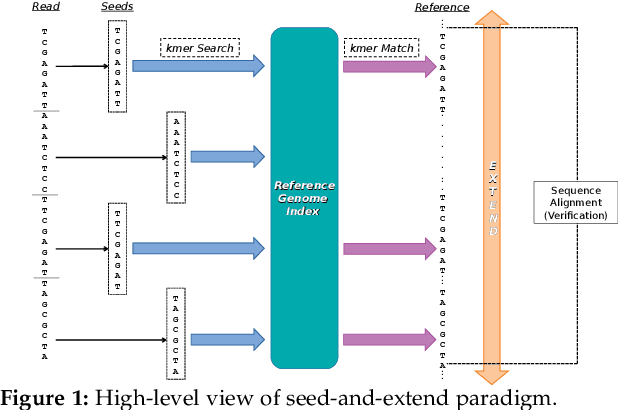
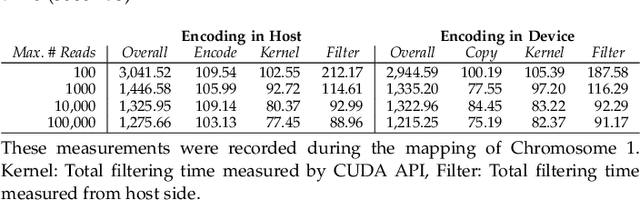

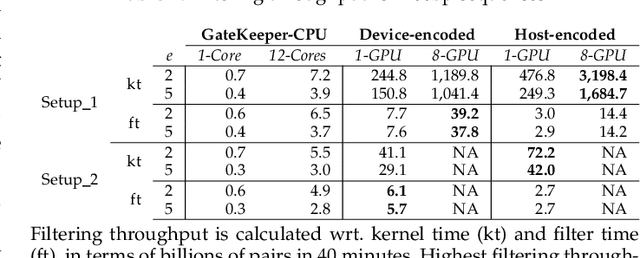
At the last step of short read mapping, the candidate locations of the reads on the reference genome are verified to compute their differences from the corresponding reference segments using sequence alignment algorithms. Calculating the similarities and differences between two sequences is still computationally expensive since approximate string matching techniques traditionally inherit dynamic programming algorithms with quadratic time and space complexity. We introduce GateKeeper-GPU, a fast and accurate pre-alignment filter that efficiently reduces the need for expensive sequence alignment. GateKeeper-GPU provides two main contributions: first, improving the filtering accuracy of GateKeeper(state-of-the-art lightweight pre-alignment filter), second, exploiting the massive parallelism provided by the large number of GPU threads of modern GPUs to examine numerous sequence pairs rapidly and concurrently. GateKeeper-GPU accelerates the sequence alignment by up to 2.9x and provides up to 1.4x speedup to the end-to-end execution time of a comprehensive read mapper (mrFAST). GateKeeper-GPU is available at https://github.com/BilkentCompGen/GateKeeper-GPU