 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeToxicBlend: Virtual Screening of Toxic Compounds with Ensemble Predictors
Jun 12, 2018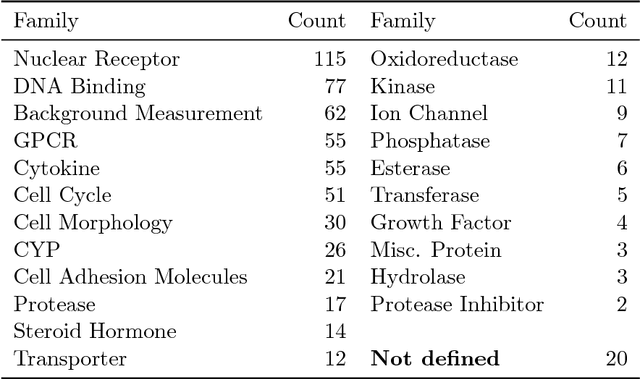
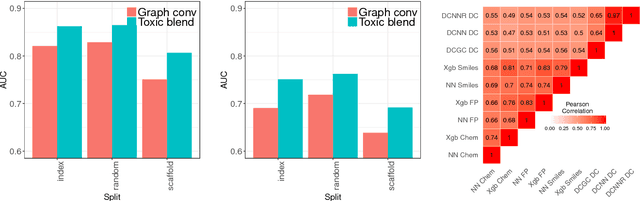
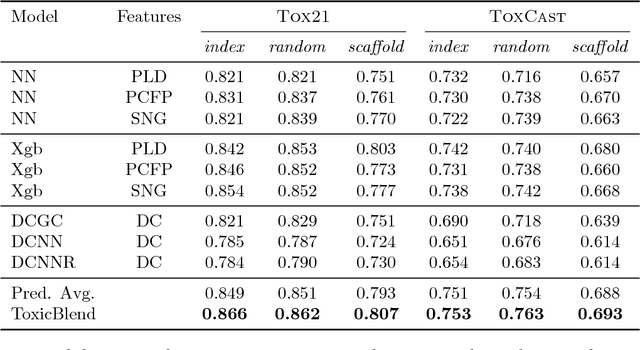
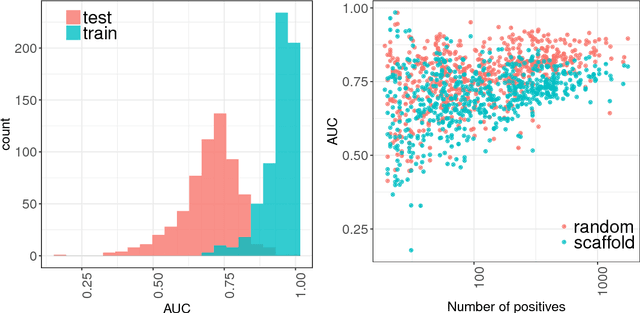
Timely assessment of compound toxicity is one of the biggest challenges facing the pharmaceutical industry today. A significant proportion of compounds identified as potential leads are ultimately discarded due to the toxicity they induce. In this paper, we propose a novel machine learning approach for the prediction of molecular activity on ToxCast targets. We combine extreme gradient boosting with fully-connected and graph-convolutional neural network architectures trained on QSAR physical molecular property descriptors, PubChem molecular fingerprints, and SMILES sequences. Our ensemble predictor leverages the strengths of each individual technique, significantly outperforming existing state-of-the art models on the ToxCast and Tox21 toxicity-prediction datasets. We provide free access to molecule toxicity prediction using our model at http://www.owkin.com/toxicblend.
Robust Detection of Covariate-Treatment Interactions in Clinical Trials
Dec 21, 2017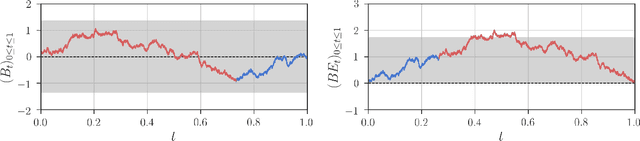

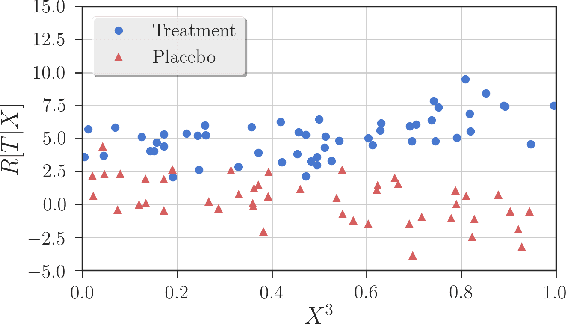
Detection of interactions between treatment effects and patient descriptors in clinical trials is critical for optimizing the drug development process. The increasing volume of data accumulated in clinical trials provides a unique opportunity to discover new biomarkers and further the goal of personalized medicine, but it also requires innovative robust biomarker detection methods capable of detecting non-linear, and sometimes weak, signals. We propose a set of novel univariate statistical tests, based on the theory of random walks, which are able to capture non-linear and non-monotonic covariate-treatment interactions. We also propose a novel combined test, which leverages the power of all of our proposed univariate tests into a single general-case tool. We present results for both synthetic trials as well as real-world clinical trials, where we compare our method with state-of-the-art techniques and demonstrate the utility and robustness of our approach.
Many-to-Many Graph Matching: a Continuous Relaxation Approach
Apr 28, 2010



Graphs provide an efficient tool for object representation in various computer vision applications. Once graph-based representations are constructed, an important question is how to compare graphs. This problem is often formulated as a graph matching problem where one seeks a mapping between vertices of two graphs which optimally aligns their structure. In the classical formulation of graph matching, only one-to-one correspondences between vertices are considered. However, in many applications, graphs cannot be matched perfectly and it is more interesting to consider many-to-many correspondences where clusters of vertices in one graph are matched to clusters of vertices in the other graph. In this paper, we formulate the many-to-many graph matching problem as a discrete optimization problem and propose an approximate algorithm based on a continuous relaxation of the combinatorial problem. We compare our method with other existing methods on several benchmark computer vision datasets.
A new protein binding pocket similarity measure based on comparison of 3D atom clouds: application to ligand prediction
Jul 09, 2009


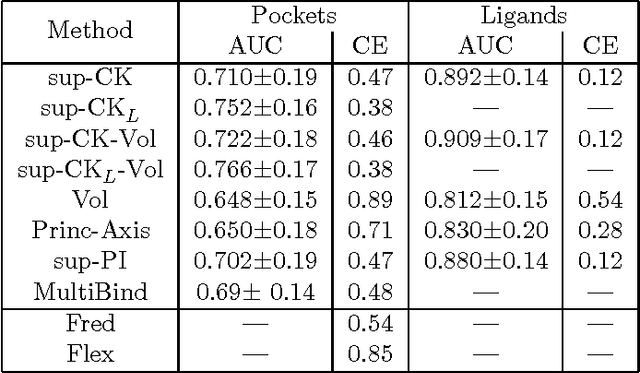
Motivation: Prediction of ligands for proteins of known 3D structure is important to understand structure-function relationship, predict molecular function, or design new drugs. Results: We explore a new approach for ligand prediction in which binding pockets are represented by atom clouds. Each target pocket is compared to an ensemble of pockets of known ligands. Pockets are aligned in 3D space with further use of convolution kernels between clouds of points. Performance of the new method for ligand prediction is compared to those of other available measures and to docking programs. We discuss two criteria to compare the quality of similarity measures: area under ROC curve (AUC) and classification based scores. We show that the latter is better suited to evaluate the methods with respect to ligand prediction. Our results on existing and new benchmarks indicate that the new method outperforms other approaches, including docking. Availability: The new method is available at http://cbio.ensmp.fr/paris/ Contact: mikhail.zaslavskiy@mines-paristech.fr
A path following algorithm for the graph matching problem
Oct 27, 2008



We propose a convex-concave programming approach for the labeled weighted graph matching problem. The convex-concave programming formulation is obtained by rewriting the weighted graph matching problem as a least-square problem on the set of permutation matrices and relaxing it to two different optimization problems: a quadratic convex and a quadratic concave optimization problem on the set of doubly stochastic matrices. The concave relaxation has the same global minimum as the initial graph matching problem, but the search for its global minimum is also a hard combinatorial problem. We therefore construct an approximation of the concave problem solution by following a solution path of a convex-concave problem obtained by linear interpolation of the convex and concave formulations, starting from the convex relaxation. This method allows to easily integrate the information on graph label similarities into the optimization problem, and therefore to perform labeled weighted graph matching. The algorithm is compared with some of the best performing graph matching methods on four datasets: simulated graphs, QAPLib, retina vessel images and handwritten chinese characters. In all cases, the results are competitive with the state-of-the-art.