 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePhaGO: Protein function annotation for bacteriophages by integrating the genomic context
Aug 12, 2024



Bacteriophages are viruses that target bacteria, playing a crucial role in microbial ecology. Phage proteins are important in understanding phage biology, such as virus infection, replication, and evolution. Although a large number of new phages have been identified via metagenomic sequencing, many of them have limited protein function annotation. Accurate function annotation of phage proteins presents several challenges, including their inherent diversity and the scarcity of annotated ones. Existing tools have yet to fully leverage the unique properties of phages in annotating protein functions. In this work, we propose a new protein function annotation tool for phages by leveraging the modular genomic structure of phage genomes. By employing embeddings from the latest protein foundation models and Transformer to capture contextual information between proteins in phage genomes, PhaGO surpasses state-of-the-art methods in annotating diverged proteins and proteins with uncommon functions by 6.78% and 13.05% improvement, respectively. PhaGO can annotate proteins lacking homology search results, which is critical for characterizing the rapidly accumulating phage genomes. We demonstrate the utility of PhaGO by identifying 688 potential holins in phages, which exhibit high structural conservation with known holins. The results show the potential of PhaGO to extend our understanding of newly discovered phages.
EDU-level Extractive Summarization with Varying Summary Lengths
Oct 08, 2022
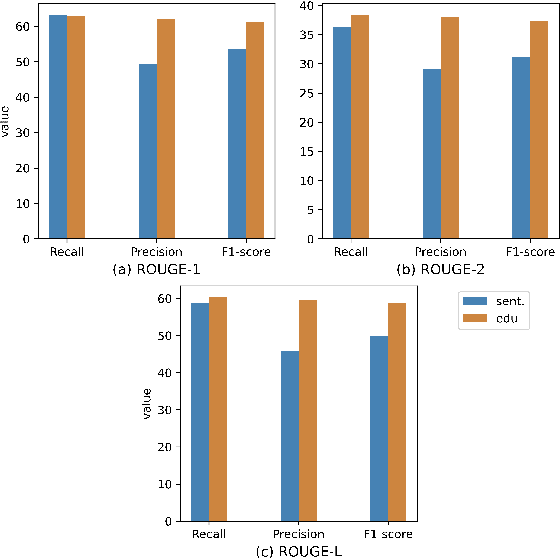
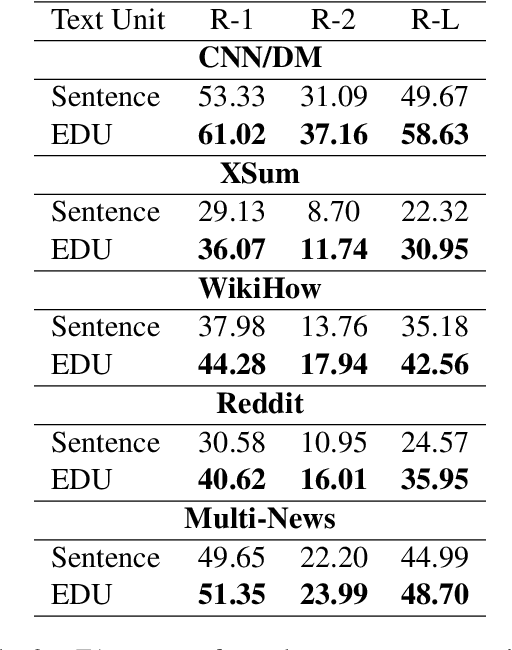
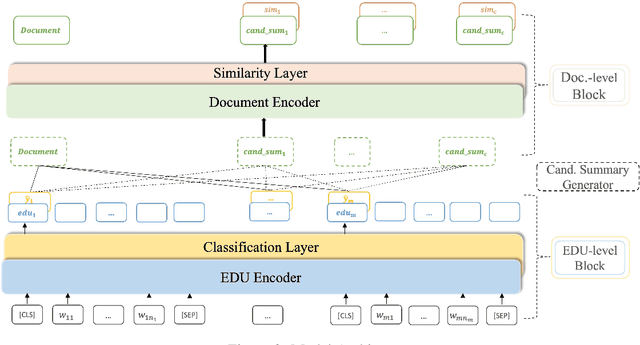
Extractive models usually formulate text summarization as extracting top-k important sentences from document as summary. Few work exploited extracting finer-grained Elementary Discourse Unit (EDU) and there is little analysis and justification for the extractive unit selection. To fill such a gap, this paper firstly conducts oracle analysis to compare the upper bound of performance for models based on EDUs and sentences. The analysis provides evidences from both theoretical and experimental perspectives to justify that EDUs make more concise and precise summary than sentences without losing salient information. Then, considering this merit of EDUs, this paper further proposes EDU-level extractive model with Varying summary Lengths (EDU-VL) and develops the corresponding learning algorithm. EDU-VL learns to encode and predict probabilities of EDUs in document, and encode EDU-level candidate summaries with different lengths based on various $k$ values and select the best candidate summary in an end-to-end training manner. Finally, the proposed and developed approach is experimented on single and multi-document benchmark datasets and shows the improved performances in comparison with the state-of-the-art models.
CHERRY: a Computational metHod for accuratE pRediction of virus-pRokarYotic interactions using a graph encoder-decoder model
Jan 04, 2022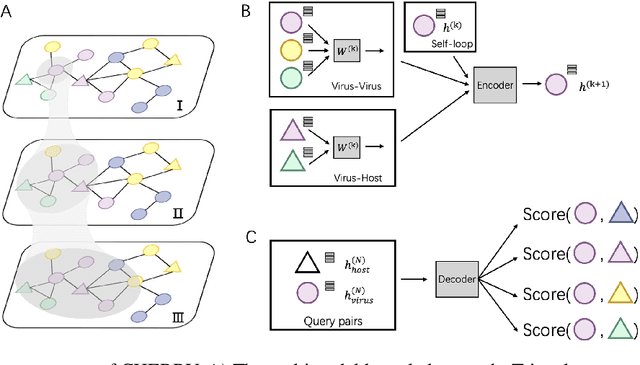
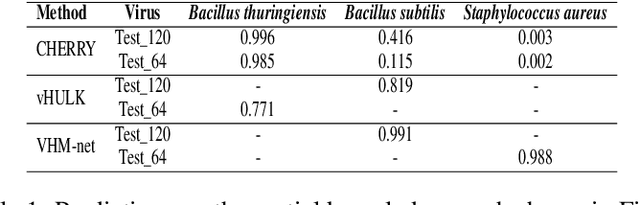
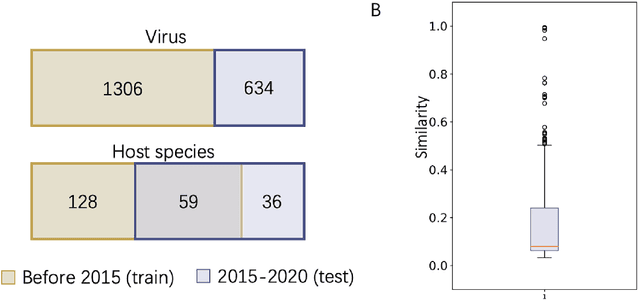
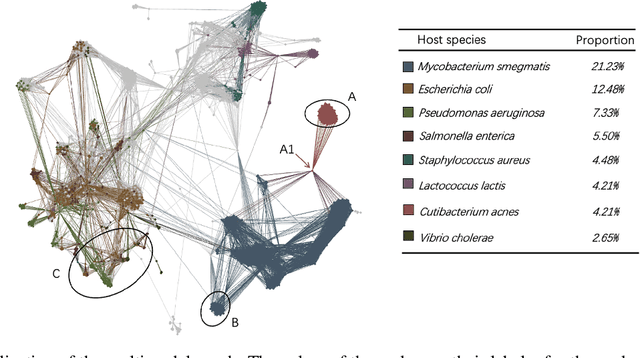
Prokaryotic viruses, which infect bacteria and archaea, are key players in microbial communities. Predicting the hosts of prokaryotic viruses helps decipher the dynamic relationship between microbes. Although there are experimental methods for host identification, they are either labor-intensive or require the cultivation of the host cells, creating a need for computational host prediction. Despite some promising results, computational host prediction remains a challenge because of the limited known interactions and the sheer amount of sequenced phages by high-throughput sequencing technologies. The state-of-the-art methods can only achieve 43% accuracy at the species level. This work presents CHERRY, a tool formulating host prediction as link prediction in a knowledge graph. As a virus-prokaryotic interaction prediction tool, CHERRY can be applied to predict hosts for newly discovered viruses and also the viruses infecting antibiotic-resistant bacteria. We demonstrated the utility of CHERRY for both applications and compared its performance with the state-of-the-art methods in different scenarios. To our best knowledge, CHERRY has the highest accuracy in identifying virus-prokaryote interactions. It outperforms all the existing methods at the species level with an accuracy increase of 37%. In addition, CHERRY's performance is more stable on short contigs than other tools.
Detecting the hosts of bacteriophages using GCN-based semi-supervised learning
May 28, 2021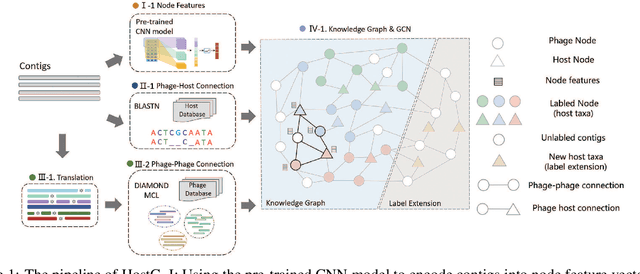
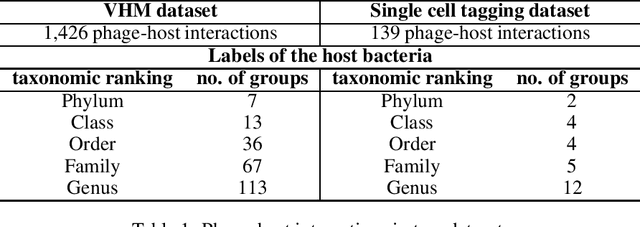
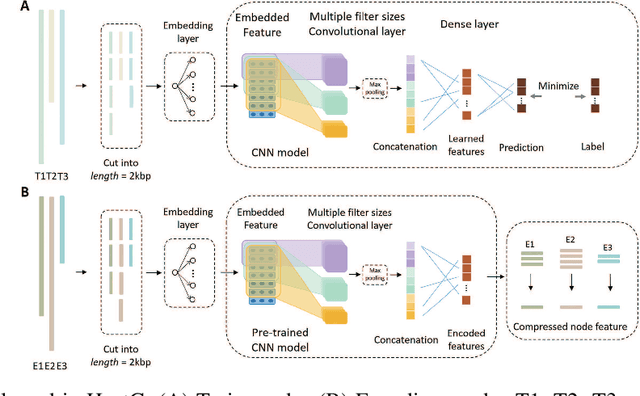
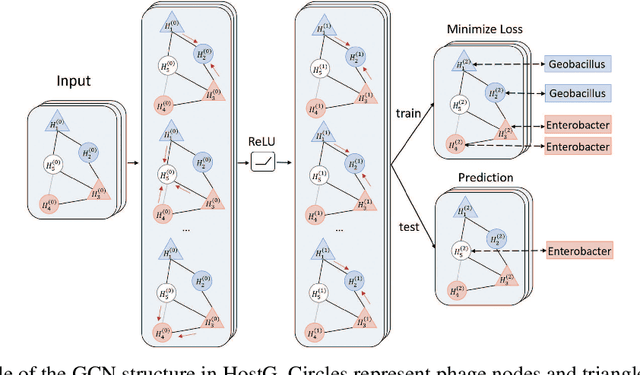
Motivation: Bacteriophages (aka phages) are viruses that infect bacteria and archaea. Thus, they play important regulatory roles in natural and host-associated ecosystems. As the most abundant and diverse biological entities in the biosphere, phages have received increased attention in their research and applications. In particular, identifying their hosts provides key knowledge for their usages as antibiotics. High-throughput sequencing and its application to the microbiome have offered new opportunities for phage host detection. However, there are two main challenges for computational host prediction. First, the known phage-host relationships are very limited compared to sequenced phages. Second, although the sequence similarity between phages and bacteria has been used as a major feature for host prediction, the alignment is either missing or ambiguous for accurate host prediction. Thus, there is still a need to improve the accuracy of host prediction. Results: In this work, we present a semi-supervised learning model, named HostG, to conduct host prediction for novel phages. We construct a knowledge graph by utilizing both phage-phage protein similarity and phage-host DNA sequence similarity. Then graph convolutional network (GCN) is adopted to exploit phages with or without known hosts in training to enhance the learning ability. During the GCN training, we minimize the expected calibrated error (ECE) to ensure the confidence of the predictions. We tested HostG on both simulated and real sequencing data and the results demonstrated that it competes favorably against the state-of-the-art pipelines.
Bacteriophage classification for assembled contigs using Graph Convolutional Network
Feb 07, 2021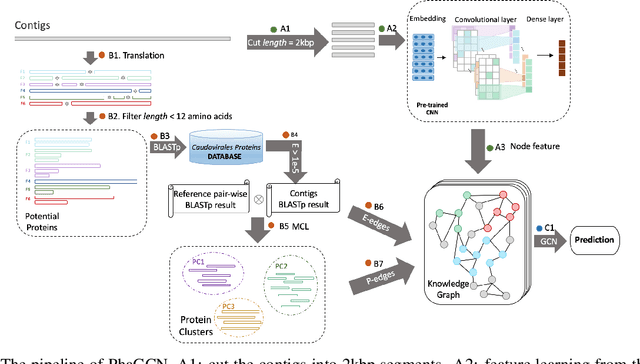
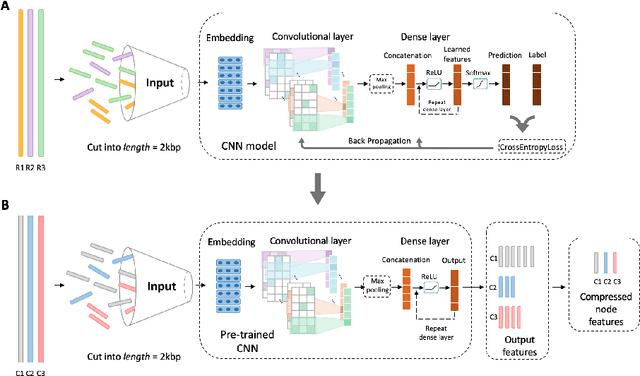
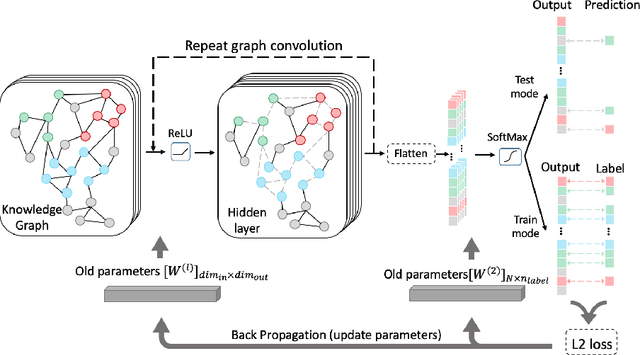
Motivation: Bacteriophages (aka phages), which mainly infect bacteria, play key roles in the biology of microbes. As the most abundant biological entities on the planet, the number of discovered phages is only the tip of the iceberg. Recently, many new phages have been revealed using high throughput sequencing, particularly metagenomic sequencing. Compared to the fast accumulation of phage-like sequences, there is a serious lag in taxonomic classification of phages. High diversity, abundance, and limited known phages pose great challenges for taxonomic analysis. In particular, alignment-based tools have difficulty in classifying fast accumulating contigs assembled from metagenomic data. Results: In this work, we present a novel semi-supervised learning model, named PhaGCN, to conduct taxonomic classification for phage contigs. In this learning model, we construct a knowledge graph by combining the DNA sequence features learned by convolutional neural network (CNN) and protein sequence similarity gained from gene-sharing network. Then we apply graph convolutional network (GCN) to utilize both the labeled and unlabeled samples in training to enhance the learning ability. We tested PhaGCN on both simulated and real sequencing data. The results clearly show that our method competes favorably against available phage classification tools.
ProDOMA: improve PROtein DOMAin classification for third-generation sequencing reads using deep learning
Sep 26, 2020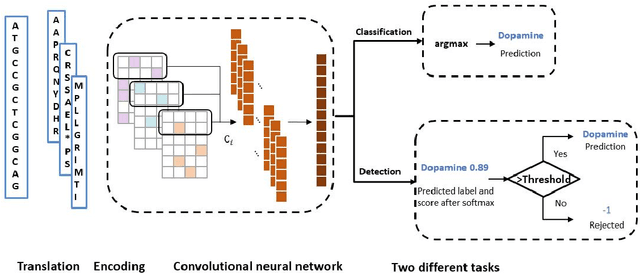
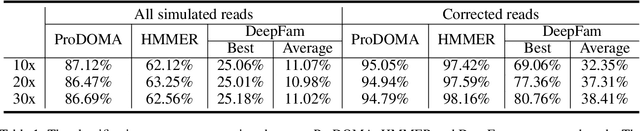
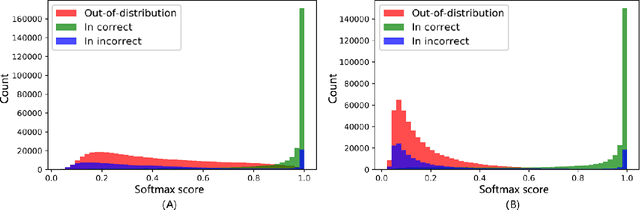

Motivation: With the development of third-generation sequencing technologies, people are able to obtain DNA sequences with lengths from 10s to 100s of kb. These long reads allow protein domain annotation without assembly, thus can produce important insights into the biological functions of the underlying data. However, the high error rate in third-generation sequencing data raises a new challenge to established domain analysis pipelines. The state-of-the-art methods are not optimized for noisy reads and have shown unsatisfactory accuracy of domain classification in third-generation sequencing data. New computational methods are still needed to improve the performance of domain prediction in long noisy reads. Results: In this work, we introduce ProDOMA, a deep learning model that conducts domain classification for third-generation sequencing reads. It uses deep neural networks with 3-frame translation encoding to learn conserved features from partially correct translations. In addition, we formulate our problem as an open-set problem and thus our model can reject unrelated DNA reads such as those from noncoding regions. In the experiments on simulated reads of protein coding sequences and real reads from the human genome, our model outperforms HMMER and DeepFam on protein domain classification. In summary, ProDOMA is a useful end-to-end protein domain analysis tool for long noisy reads without relying on error correction. Availability: The source code and the trained model are freely available at https://github.com/strideradu/ProDOMA. Contact: yannisun@cityu.edu.hk