 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeExplaining models relating objects and privacy
May 02, 2024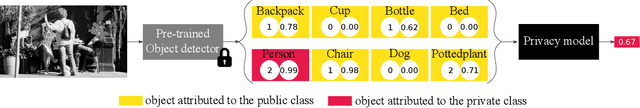
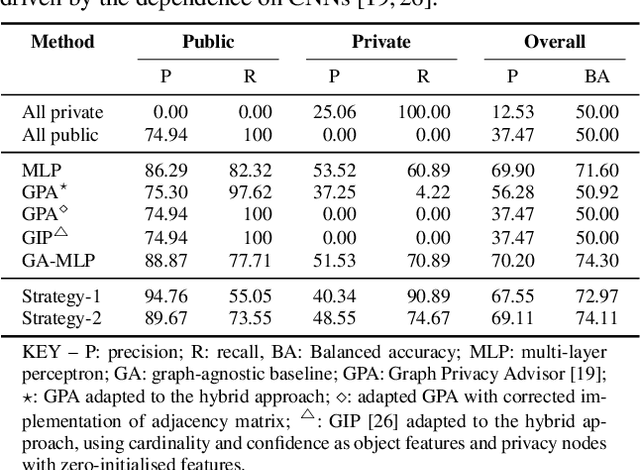
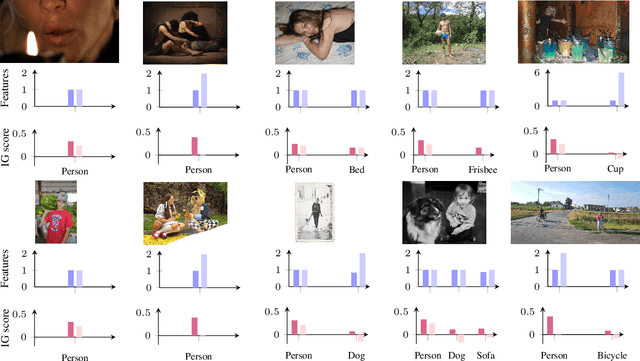
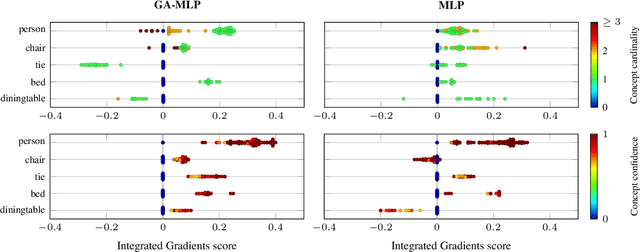
Accurately predicting whether an image is private before sharing it online is difficult due to the vast variety of content and the subjective nature of privacy itself. In this paper, we evaluate privacy models that use objects extracted from an image to determine why the image is predicted as private. To explain the decision of these models, we use feature-attribution to identify and quantify which objects (and which of their features) are more relevant to privacy classification with respect to a reference input (i.e., no objects localised in an image) predicted as public. We show that the presence of the person category and its cardinality is the main factor for the privacy decision. Therefore, these models mostly fail to identify private images depicting documents with sensitive data, vehicle ownership, and internet activity, or public images with people (e.g., an outdoor concert or people walking in a public space next to a famous landmark). As baselines for future benchmarks, we also devise two strategies that are based on the person presence and cardinality and achieve comparable classification performance of the privacy models.
A Comparative Analysis of Gene Expression Profiling by Statistical and Machine Learning Approaches
Feb 01, 2024



Many machine learning models have been proposed to classify phenotypes from gene expression data. In addition to their good performance, these models can potentially provide some understanding of phenotypes by extracting explanations for their decisions. These explanations often take the form of a list of genes ranked in order of importance for the predictions, the highest-ranked genes being interpreted as linked to the phenotype. We discuss the biological and the methodological limitations of such explanations. Experiments are performed on several datasets gathering cancer and healthy tissue samples from the TCGA, GTEx and TARGET databases. A collection of machine learning models including logistic regression, multilayer perceptron, and graph neural network are trained to classify samples according to their cancer type. Gene rankings are obtained from explainability methods adapted to these models, and compared to the ones from classical statistical feature selection methods such as mutual information, DESeq2, and EdgeR. Interestingly, on simple tasks, we observe that the information learned by black-box neural networks is related to the notion of differential expression. In all cases, a small set containing the best-ranked genes is sufficient to achieve a good classification. However, these genes differ significantly between the methods and similar classification performance can be achieved with numerous lower ranked genes. In conclusion, although these methods enable the identification of biomarkers characteristic of certain pathologies, our results question the completeness of the selected gene sets and thus of explainability by the identification of the underlying biological processes.
Studying Limits of Explainability by Integrated Gradients for Gene Expression Models
Mar 19, 2023


Understanding the molecular processes that drive cellular life is a fundamental question in biological research. Ambitious programs have gathered a number of molecular datasets on large populations. To decipher the complex cellular interactions, recent work has turned to supervised machine learning methods. The scientific questions are formulated as classical learning problems on tabular data or on graphs, e.g. phenotype prediction from gene expression data. In these works, the input features on which the individual predictions are predominantly based are often interpreted as indicative of the cause of the phenotype, such as cancer identification. Here, we propose to explore the relevance of the biomarkers identified by Integrated Gradients, an explainability method for feature attribution in machine learning. Through a motivating example on The Cancer Genome Atlas, we show that ranking features by importance is not enough to robustly identify biomarkers. As it is difficult to evaluate whether biomarkers reflect relevant causes without known ground truth, we simulate gene expression data by proposing a hierarchical model based on Latent Dirichlet Allocation models. We also highlight good practices for evaluating explanations for genomics data and propose a direction to derive more insights from these explanations.