 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeRxRx1: A Dataset for Evaluating Experimental Batch Correction Methods
Jan 13, 2023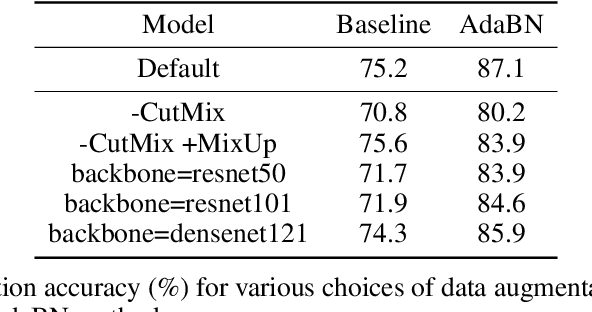



High-throughput screening techniques are commonly used to obtain large quantities of data in many fields of biology. It is well known that artifacts arising from variability in the technical execution of different experimental batches within such screens confound these observations and can lead to invalid biological conclusions. It is therefore necessary to account for these batch effects when analyzing outcomes. In this paper we describe RxRx1, a biological dataset designed specifically for the systematic study of batch effect correction methods. The dataset consists of 125,510 high-resolution fluorescence microscopy images of human cells under 1,138 genetic perturbations in 51 experimental batches across 4 cell types. Visual inspection of the images alone clearly demonstrates significant batch effects. We propose a classification task designed to evaluate the effectiveness of experimental batch correction methods on these images and examine the performance of a number of correction methods on this task. Our goal in releasing RxRx1 is to encourage the development of effective experimental batch correction methods that generalize well to unseen experimental batches. The dataset can be downloaded at https://rxrx.ai.
METCC: METric learning for Confounder Control Making distance matter in high dimensional biological analysis
Dec 07, 2018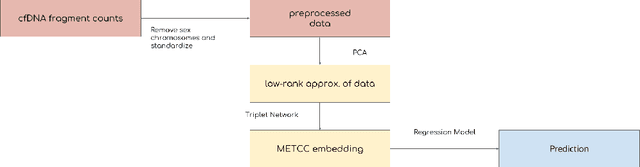
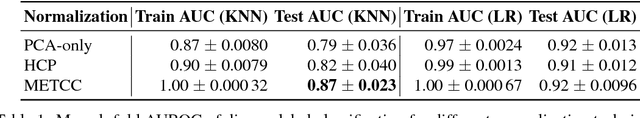
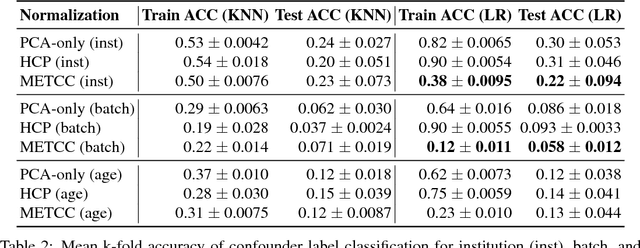
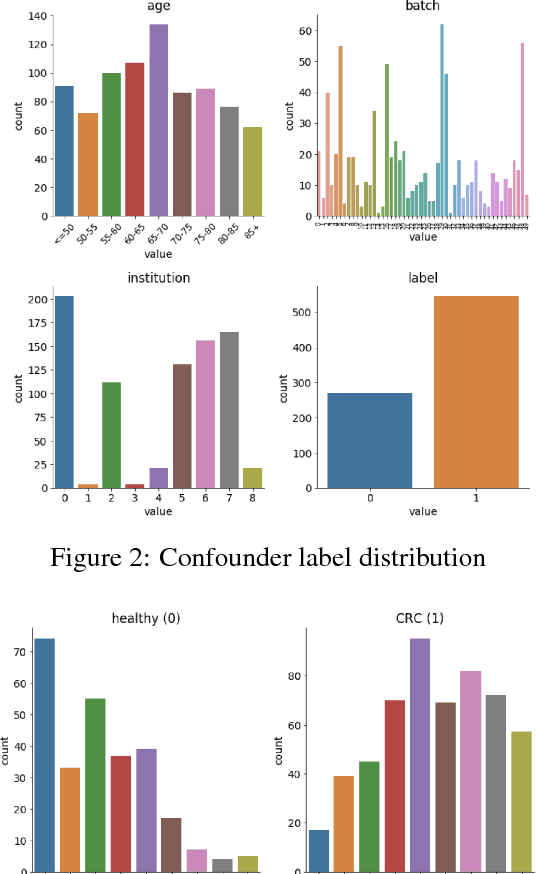
High-dimensional data acquired from biological experiments such as next generation sequencing are subject to a number of confounding effects. These effects include both technical effects, such as variation across batches from instrument noise or sample processing, or institution-specific differences in sample acquisition and physical handling, as well as biological effects arising from true but irrelevant differences in the biology of each sample, such as age biases in diseases. Prior work has used linear methods to adjust for such batch effects. Here, we apply contrastive metric learning by a non-linear triplet network to optimize the ability to distinguish biologically distinct sample classes in the presence of irrelevant technical and biological variation. Using whole-genome cell-free DNA data from 817 patients, we demonstrate that our approach, METric learning for Confounder Control (METCC), is able to match or exceed the classification performance achieved using a best-in-class linear method (HCP) or no normalization. Critically, results from METCC appear less confounded by irrelevant technical variables like institution and batch than those from other methods even without access to high quality metadata information required by many existing techniques; offering hope for improved generalization.