 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeIntroducing δ-XAI: a novel sensitivity-based method for local AI explanations
Jul 29, 2024
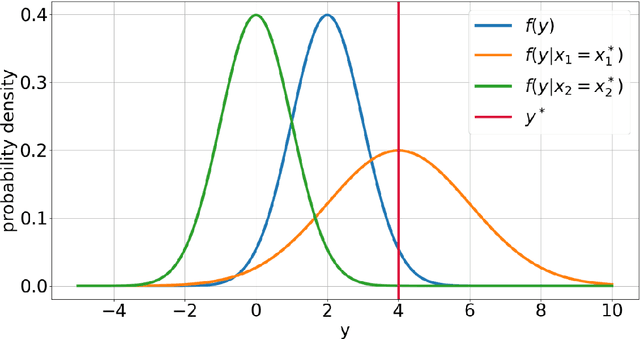

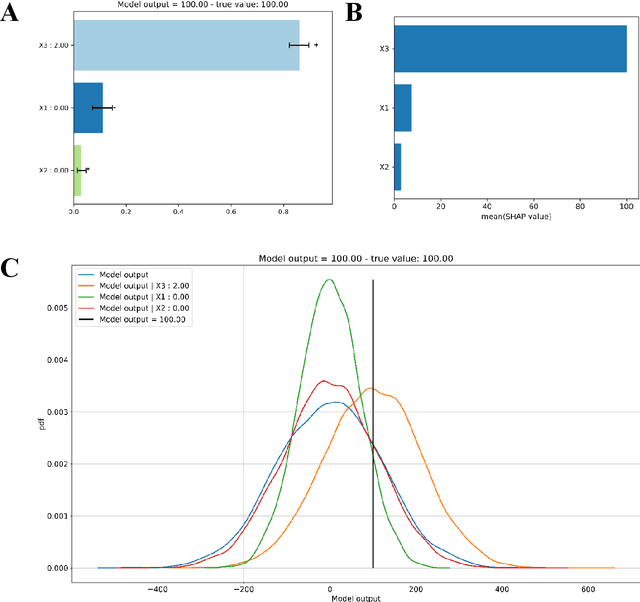
Explainable Artificial Intelligence (XAI) is central to the debate on integrating Artificial Intelligence (AI) and Machine Learning (ML) algorithms into clinical practice. High-performing AI/ML models, such as ensemble learners and deep neural networks, often lack interpretability, hampering clinicians' trust in their predictions. To address this, XAI techniques are being developed to describe AI/ML predictions in human-understandable terms. One promising direction is the adaptation of sensitivity analysis (SA) and global sensitivity analysis (GSA), which inherently rank model inputs by their impact on predictions. Here, we introduce a novel delta-XAI method that provides local explanations of ML model predictions by extending the delta index, a GSA metric. The delta-XAI index assesses the impact of each feature's value on the predicted output for individual instances in both regression and classification problems. We formalize the delta-XAI index and provide code for its implementation. The delta-XAI method was evaluated on simulated scenarios using linear regression models, with Shapley values serving as a benchmark. Results showed that the delta-XAI index is generally consistent with Shapley values, with notable discrepancies in models with highly impactful or extreme feature values. The delta-XAI index demonstrated higher sensitivity in detecting dominant features and handling extreme feature values. Qualitatively, the delta-XAI provides intuitive explanations by leveraging probability density functions, making feature rankings clearer and more explainable for practitioners. Overall, the delta-XAI method appears promising for robustly obtaining local explanations of ML model predictions. Further investigations in real-world clinical settings will be conducted to evaluate its impact on AI-assisted clinical workflows.
Evaluation of Predictive Reliability to Foster Trust in Artificial Intelligence. A case study in Multiple Sclerosis
Feb 27, 2024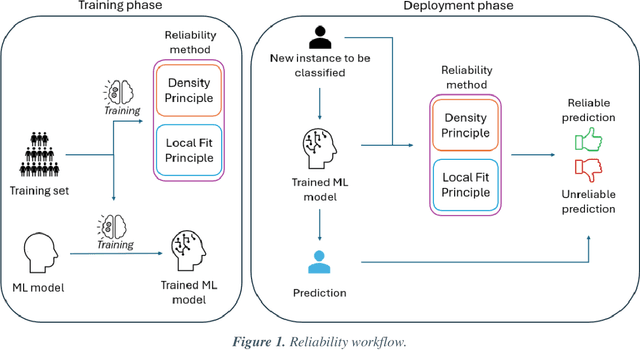

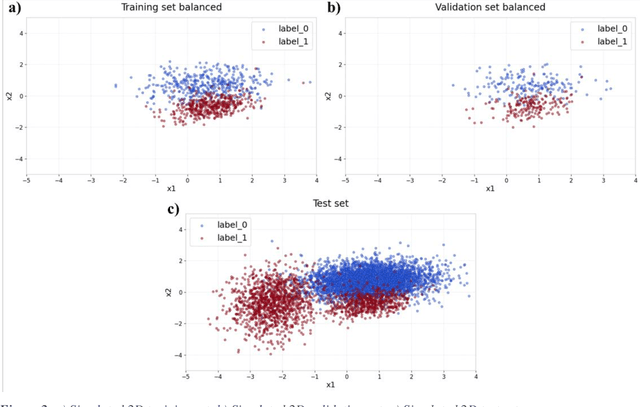
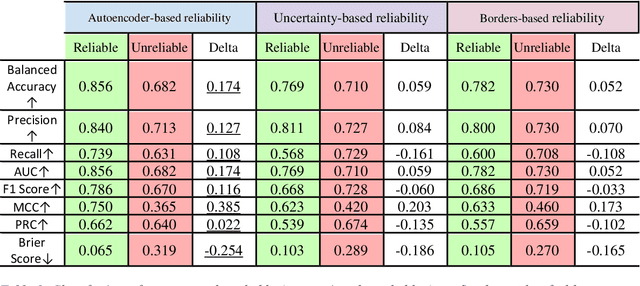
Applying Artificial Intelligence (AI) and Machine Learning (ML) in critical contexts, such as medicine, requires the implementation of safety measures to reduce risks of harm in case of prediction errors. Spotting ML failures is of paramount importance when ML predictions are used to drive clinical decisions. ML predictive reliability measures the degree of trust of a ML prediction on a new instance, thus allowing decision-makers to accept or reject it based on its reliability. To assess reliability, we propose a method that implements two principles. First, our approach evaluates whether an instance to be classified is coming from the same distribution of the training set. To do this, we leverage Autoencoders (AEs) ability to reconstruct the training set with low error. An instance is considered Out-of-Distribution (OOD) if the AE reconstructs it with a high error. Second, it is evaluated whether the ML classifier has good performances on samples similar to the newly classified instance by using a proxy model. We show that this approach is able to assess reliability both in a simulated scenario and on a model trained to predict disease progression of Multiple Sclerosis patients. We also developed a Python package, named relAI, to embed reliability measures into ML pipelines. We propose a simple approach that can be used in the deployment phase of any ML model to suggest whether to trust predictions or not. Our method holds the promise to provide effective support to clinicians by spotting potential ML failures during deployment.
Tree-based local explanations of machine learning model predictions, AraucanaXAI
Oct 15, 2021Increasingly complex learning methods such as boosting, bagging and deep learning have made ML models more accurate, but harder to understand and interpret. A tradeoff between performance and intelligibility is often to be faced, especially in high-stakes applications like medicine. In the present article we propose a novel methodological approach for generating explanations of the predictions of a generic ML model, given a specific instance for which the prediction has been made, that can tackle both classification and regression tasks. Advantages of the proposed XAI approach include improved fidelity to the original model, the ability to deal with non-linear decision boundaries, and native support to both classification and regression problems
The Gene Mover's Distance: Single-cell similarity via Optimal Transport
Feb 01, 2021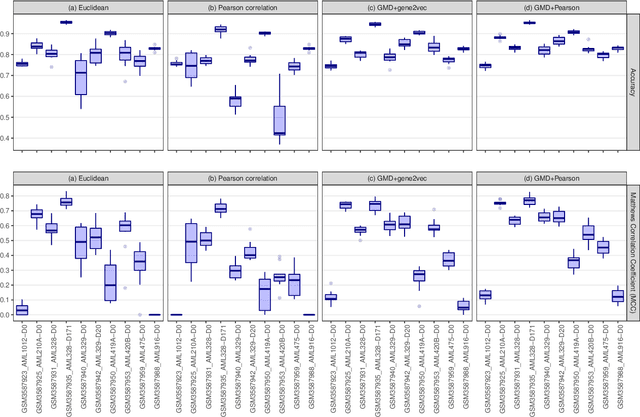
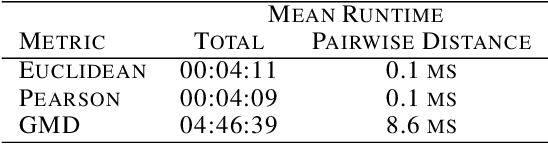
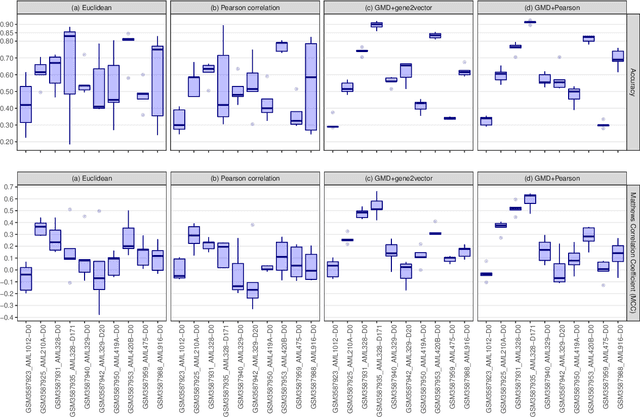
This paper introduces the Gene Mover's Distance, a measure of similarity between a pair of cells based on their gene expression profiles obtained via single-cell RNA sequencing. The underlying idea of the proposed distance is to interpret the gene expression array of a single cell as a discrete probability measure. The distance between two cells is hence computed by solving an Optimal Transport problem between the two corresponding discrete measures. In the Optimal Transport model, we use two types of cost function for measuring the distance between a pair of genes. The first cost function exploits a gene embedding, called gene2vec, which is used to map each gene to a high dimensional vector: the cost of moving a unit of mass of gene expression from a gene to another is set to the Euclidean distance between the corresponding embedded vectors. The second cost function is based on a Pearson distance among pairs of genes. In both cost functions, the more two genes are correlated, the lower is their distance. We exploit the Gene Mover's Distance to solve two classification problems: the classification of cells according to their condition and according to their type. To assess the impact of our new metric, we compare the performances of a $k$-Nearest Neighbor classifier using different distances. The computational results show that the Gene Mover's Distance is competitive with the state-of-the-art distances used in the literature.