 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeFused Gromov-Wasserstein Contrastive Learning for Effective Enzyme-Reaction Screening
Dec 09, 2025Enzymes are crucial catalysts that enable a wide range of biochemical reactions. Efficiently identifying specific enzymes from vast protein libraries is essential for advancing biocatalysis. Traditional computational methods for enzyme screening and retrieval are time-consuming and resource-intensive. Recently, deep learning approaches have shown promise. However, these methods focus solely on the interaction between enzymes and reactions, overlooking the inherent hierarchical relationships within each domain. To address these limitations, we introduce FGW-CLIP, a novel contrastive learning framework based on optimizing the fused Gromov-Wasserstein distance. FGW-CLIP incorporates multiple alignments, including inter-domain alignment between reactions and enzymes and intra-domain alignment within enzymes and reactions. By introducing a tailored regularization term, our method minimizes the Gromov-Wasserstein distance between enzyme and reaction spaces, which enhances information integration across these domains. Extensive evaluations demonstrate the superiority of FGW-CLIP in challenging enzyme-reaction tasks. On the widely-used EnzymeMap benchmark, FGW-CLIP achieves state-of-the-art performance in enzyme virtual screening, as measured by BEDROC and EF metrics. Moreover, FGW-CLIP consistently outperforms across all three splits of ReactZyme, the largest enzyme-reaction benchmark, demonstrating robust generalization to novel enzymes and reactions. These results position FGW-CLIP as a promising framework for enzyme discovery in complex biochemical settings, with strong adaptability across diverse screening scenarios.
Uni-Mol Docking V2: Towards Realistic and Accurate Binding Pose Prediction
May 20, 2024



In recent years, machine learning (ML) methods have emerged as promising alternatives for molecular docking, offering the potential for high accuracy without incurring prohibitive computational costs. However, recent studies have indicated that these ML models may overfit to quantitative metrics while neglecting the physical constraints inherent in the problem. In this work, we present Uni-Mol Docking V2, which demonstrates a remarkable improvement in performance, accurately predicting the binding poses of 77+% of ligands in the PoseBusters benchmark with an RMSD value of less than 2.0 {\AA}, and 75+% passing all quality checks. This represents a significant increase from the 62% achieved by the previous Uni-Mol Docking model. Notably, our Uni-Mol Docking approach generates chemically accurate predictions, circumventing issues such as chirality inversions and steric clashes that have plagued previous ML models. Furthermore, we observe enhanced performance in terms of high-quality predictions (RMSD values of less than 1.0 {\AA} and 1.5 {\AA}) and physical soundness when Uni-Mol Docking is combined with more physics-based methods like Uni-Dock. Our results represent a significant advancement in the application of artificial intelligence for scientific research, adopting a holistic approach to ligand docking that is well-suited for industrial applications in virtual screening and drug design. The code, data and service for Uni-Mol Docking are publicly available for use and further development in https://github.com/dptech-corp/Uni-Mol.
Do Deep Learning Methods Really Perform Better in Molecular Conformation Generation?
Feb 14, 2023Molecular conformation generation (MCG) is a fundamental and important problem in drug discovery. Many traditional methods have been developed to solve the MCG problem, such as systematic searching, model-building, random searching, distance geometry, molecular dynamics, Monte Carlo methods, etc. However, they have some limitations depending on the molecular structures. Recently, there are plenty of deep learning based MCG methods, which claim they largely outperform the traditional methods. However, to our surprise, we design a simple and cheap algorithm (parameter-free) based on the traditional methods and find it is comparable to or even outperforms deep learning based MCG methods in the widely used GEOM-QM9 and GEOM-Drugs benchmarks. In particular, our design algorithm is simply the clustering of the RDKIT-generated conformations. We hope our findings can help the community to revise the deep learning methods for MCG. The code of the proposed algorithm could be found at https://gist.github.com/ZhouGengmo/5b565f51adafcd911c0bc115b2ef027c.
Predicting Protein-Ligand Binding Affinity via Joint Global-Local Interaction Modeling
Sep 18, 2022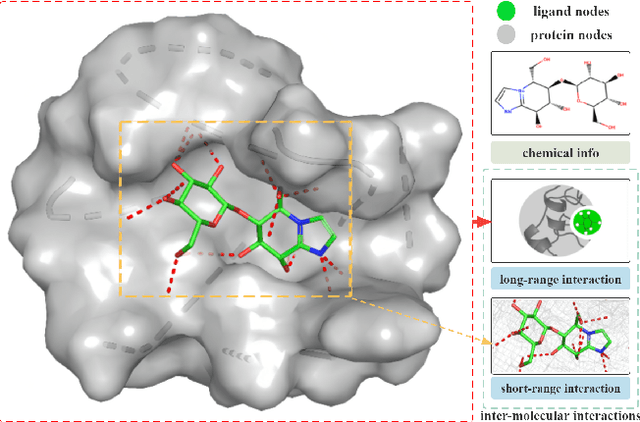
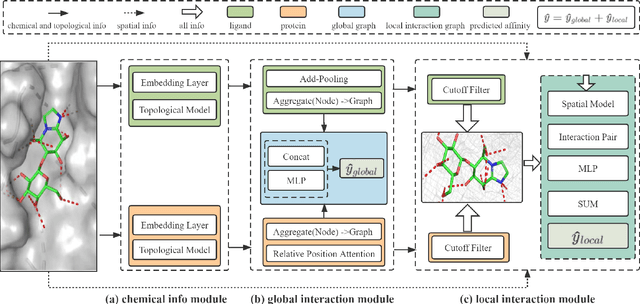
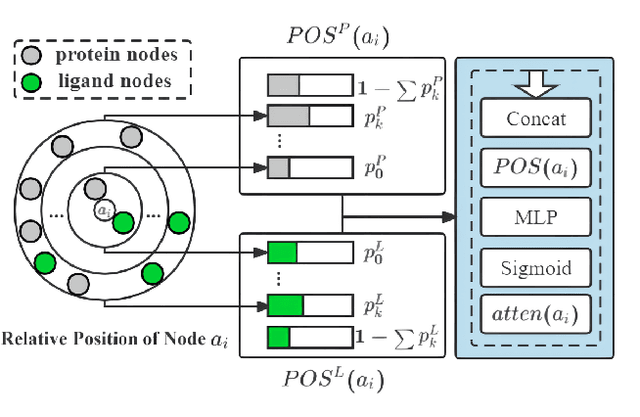
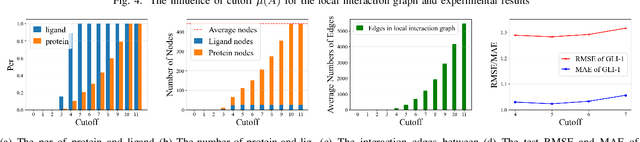
The prediction of protein-ligand binding affinity is of great significance for discovering lead compounds in drug research. Facing this challenging task, most existing prediction methods rely on the topological and/or spatial structure of molecules and the local interactions while ignoring the multi-level inter-molecular interactions between proteins and ligands, which often lead to sub-optimal performance. To solve this issue, we propose a novel global-local interaction (GLI) framework to predict protein-ligand binding affinity. In particular, our GLI framework considers the inter-molecular interactions between proteins and ligands, which involve not only the high-energy short-range interactions between closed atoms but also the low-energy long-range interactions between non-bonded atoms. For each pair of protein and ligand, our GLI embeds the long-range interactions globally and aggregates local short-range interactions, respectively. Such a joint global-local interaction modeling strategy helps to improve prediction accuracy, and the whole framework is compatible with various neural network-based modules. Experiments demonstrate that our GLI framework outperforms state-of-the-art methods with simple neural network architectures and moderate computational costs.