 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePredicting Protein-Ligand Binding Affinity via Joint Global-Local Interaction Modeling
Paper and Code
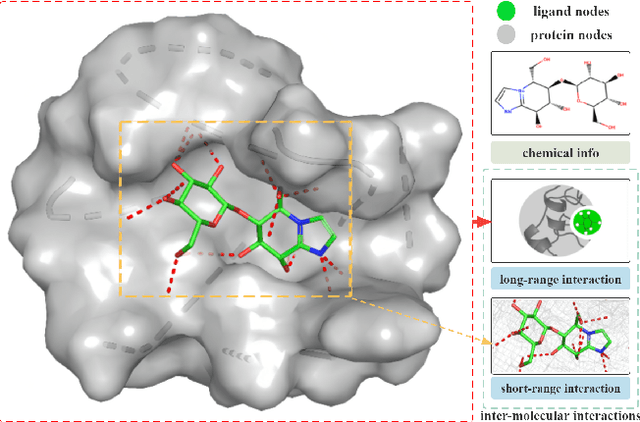
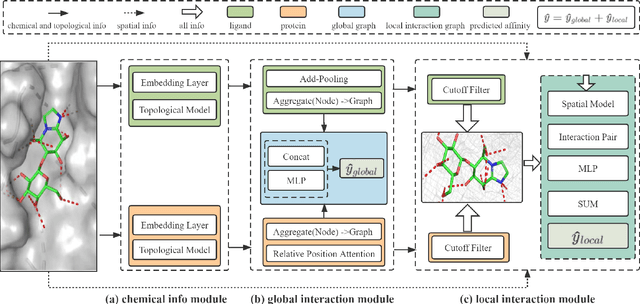
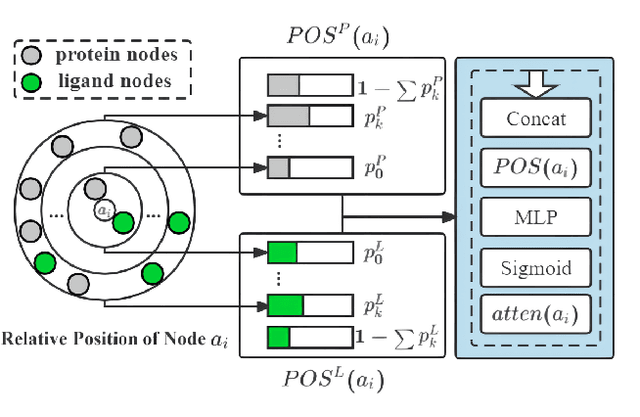
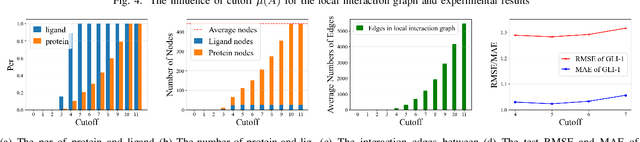
The prediction of protein-ligand binding affinity is of great significance for discovering lead compounds in drug research. Facing this challenging task, most existing prediction methods rely on the topological and/or spatial structure of molecules and the local interactions while ignoring the multi-level inter-molecular interactions between proteins and ligands, which often lead to sub-optimal performance. To solve this issue, we propose a novel global-local interaction (GLI) framework to predict protein-ligand binding affinity. In particular, our GLI framework considers the inter-molecular interactions between proteins and ligands, which involve not only the high-energy short-range interactions between closed atoms but also the low-energy long-range interactions between non-bonded atoms. For each pair of protein and ligand, our GLI embeds the long-range interactions globally and aggregates local short-range interactions, respectively. Such a joint global-local interaction modeling strategy helps to improve prediction accuracy, and the whole framework is compatible with various neural network-based modules. Experiments demonstrate that our GLI framework outperforms state-of-the-art methods with simple neural network architectures and moderate computational costs.