 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePushing the limits of unconstrained machine-learned interatomic potentials
Jan 22, 2026Machine-learned interatomic potentials (MLIPs) are increasingly used to replace computationally demanding electronic-structure calculations to model matter at the atomic scale. The most commonly used model architectures are constrained to fulfill a number of physical laws exactly, from geometric symmetries to energy conservation. Evidence is mounting that relaxing some of these constraints can be beneficial to the efficiency and (somewhat surprisingly) accuracy of MLIPs, even though care should be taken to avoid qualitative failures associated with the breaking of physical symmetries. Given the recent trend of \emph{scaling up} models to larger numbers of parameters and training samples, a very important question is how unconstrained MLIPs behave in this limit. Here we investigate this issue, showing that -- when trained on large datasets -- unconstrained models can be superior in accuracy and speed when compared to physically constrained models. We assess these models both in terms of benchmark accuracy and in terms of usability in practical scenarios, focusing on static simulation workflows such as geometry optimization and lattice dynamics. We conclude that accurate unconstrained models can be applied with confidence, especially since simple inference-time modifications can be used to recover observables that are consistent with the relevant physical symmetries.
FlashMD: long-stride, universal prediction of molecular dynamics
May 25, 2025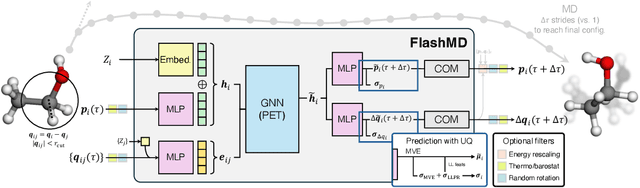
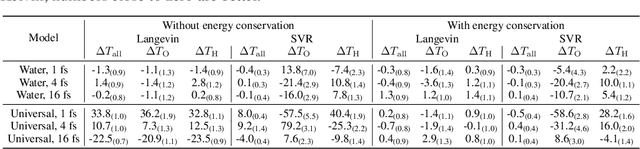
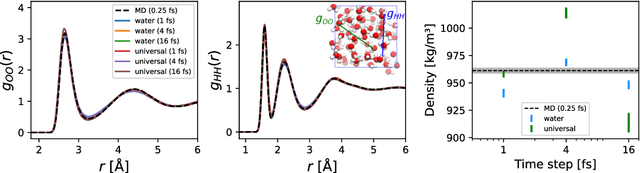
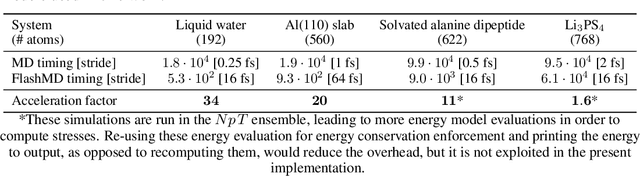
Molecular dynamics (MD) provides insights into atomic-scale processes by integrating over time the equations that describe the motion of atoms under the action of interatomic forces. Machine learning models have substantially accelerated MD by providing inexpensive predictions of the forces, but they remain constrained to minuscule time integration steps, which are required by the fast time scale of atomic motion. In this work, we propose FlashMD, a method to predict the evolution of positions and momenta over strides that are between one and two orders of magnitude longer than typical MD time steps. We incorporate considerations on the mathematical and physical properties of Hamiltonian dynamics in the architecture, generalize the approach to allow the simulation of any thermodynamic ensemble, and carefully assess the possible failure modes of such a long-stride MD approach. We validate FlashMD's accuracy in reproducing equilibrium and time-dependent properties, using both system-specific and general-purpose models, extending the ability of MD simulation to reach the long time scales needed to model microscopic processes of high scientific and technological relevance.
Representing spherical tensors with scalar-based machine-learning models
May 08, 2025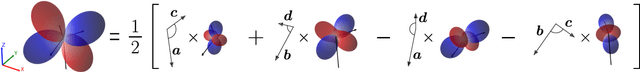
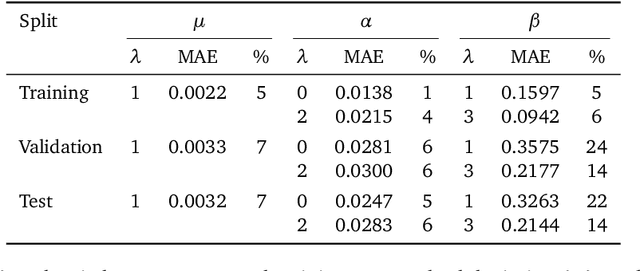
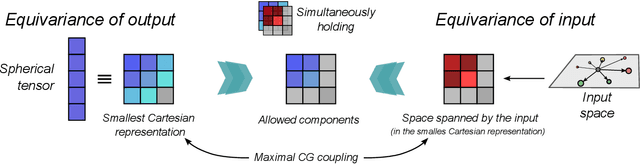
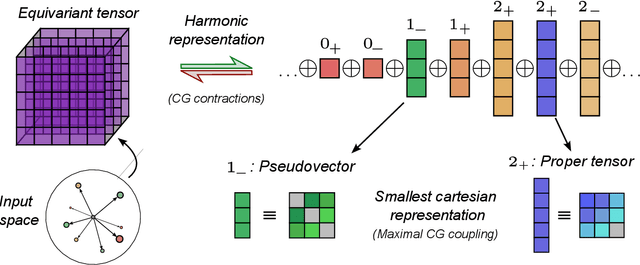
Rotational symmetry plays a central role in physics, providing an elegant framework to describe how the properties of 3D objects -- from atoms to the macroscopic scale -- transform under the action of rigid rotations. Equivariant models of 3D point clouds are able to approximate structure-property relations in a way that is fully consistent with the structure of the rotation group, by combining intermediate representations that are themselves spherical tensors. The symmetry constraints however make this approach computationally demanding and cumbersome to implement, which motivates increasingly popular unconstrained architectures that learn approximate symmetries as part of the training process. In this work, we explore a third route to tackle this learning problem, where equivariant functions are expressed as the product of a scalar function of the point cloud coordinates and a small basis of tensors with the appropriate symmetry. We also propose approximations of the general expressions that, while lacking universal approximation properties, are fast, simple to implement, and accurate in practical settings.
PET-MAD, a universal interatomic potential for advanced materials modeling
Mar 18, 2025



Machine-learning interatomic potentials (MLIPs) have greatly extended the reach of atomic-scale simulations, offering the accuracy of first-principles calculations at a fraction of the effort. Leveraging large quantum mechanical databases and expressive architectures, recent "universal" models deliver qualitative accuracy across the periodic table but are often biased toward low-energy configurations. We introduce PET-MAD, a generally applicable MLIP trained on a dataset combining stable inorganic and organic solids, systematically modified to enhance atomic diversity. Using a moderate but highly-consistent level of electronic-structure theory, we assess PET-MAD's accuracy on established benchmarks and advanced simulations of six materials. PET-MAD rivals state-of-the-art MLIPs for inorganic solids, while also being reliable for molecules, organic materials, and surfaces. It is stable and fast, enabling, out-of-the-box, the near-quantitative study of thermal and quantum mechanical fluctuations, functional properties, and phase transitions. It can be efficiently fine-tuned to deliver full quantum mechanical accuracy with a minimal number of targeted calculations.
Humanity's Last Exam
Jan 24, 2025Benchmarks are important tools for tracking the rapid advancements in large language model (LLM) capabilities. However, benchmarks are not keeping pace in difficulty: LLMs now achieve over 90\% accuracy on popular benchmarks like MMLU, limiting informed measurement of state-of-the-art LLM capabilities. In response, we introduce Humanity's Last Exam (HLE), a multi-modal benchmark at the frontier of human knowledge, designed to be the final closed-ended academic benchmark of its kind with broad subject coverage. HLE consists of 3,000 questions across dozens of subjects, including mathematics, humanities, and the natural sciences. HLE is developed globally by subject-matter experts and consists of multiple-choice and short-answer questions suitable for automated grading. Each question has a known solution that is unambiguous and easily verifiable, but cannot be quickly answered via internet retrieval. State-of-the-art LLMs demonstrate low accuracy and calibration on HLE, highlighting a significant gap between current LLM capabilities and the expert human frontier on closed-ended academic questions. To inform research and policymaking upon a clear understanding of model capabilities, we publicly release HLE at https://lastexam.ai.
The dark side of the forces: assessing non-conservative force models for atomistic machine learning
Dec 16, 2024



The use of machine learning to estimate the energy of a group of atoms, and the forces that drive them to more stable configurations, have revolutionized the fields of computational chemistry and materials discovery. In this domain, rigorous enforcement of symmetry and conservation laws has traditionally been considered essential. For this reason, interatomic forces are usually computed as the derivatives of the potential energy, ensuring energy conservation. Several recent works have questioned this physically-constrained approach, suggesting that using the forces as explicit learning targets yields a better trade-off between accuracy and computational efficiency - and that energy conservation can be learned during training. The present work investigates the applicability of such non-conservative models in microscopic simulations. We identify and demonstrate several fundamental issues, from ill-defined convergence of geometry optimization to instability in various types of molecular dynamics. Contrary to the case of rotational symmetry, lack of energy conservation is hard to learn, control, and correct. The best approach to exploit the acceleration afforded by direct force evaluation might be to use it in tandem with a conservative model, reducing - rather than eliminating - the additional cost of backpropagation, but avoiding most of the pathological behavior associated with non-conservative forces.
A prediction rigidity formalism for low-cost uncertainties in trained neural networks
Mar 04, 2024Regression methods are fundamental for scientific and technological applications. However, fitted models can be highly unreliable outside of their training domain, and hence the quantification of their uncertainty is crucial in many of their applications. Based on the solution of a constrained optimization problem, we propose "prediction rigidities" as a method to obtain uncertainties of arbitrary pre-trained regressors. We establish a strong connection between our framework and Bayesian inference, and we develop a last-layer approximation that allows the new method to be applied to neural networks. This extension affords cheap uncertainties without any modification to the neural network itself or its training procedure. We show the effectiveness of our method on a wide range of regression tasks, ranging from simple toy models to applications in chemistry and meteorology.
Wigner kernels: body-ordered equivariant machine learning without a basis
Mar 07, 2023Machine-learning models based on a point-cloud representation of a physical object are ubiquitous in scientific applications and particularly well-suited to the atomic-scale description of molecules and materials. Among the many different approaches that have been pursued, the description of local atomic environments in terms of their neighbor densities has been used widely and very succesfully. We propose a novel density-based method which involves computing ``Wigner kernels''. These are fully equivariant and body-ordered kernels that can be computed iteratively with a cost that is independent of the radial-chemical basis and grows only linearly with the maximum body-order considered. This is in marked contrast to feature-space models, which comprise an exponentially-growing number of terms with increasing order of correlations. We present several examples of the accuracy of models based on Wigner kernels in chemical applications, for both scalar and tensorial targets, reaching state-of-the-art accuracy on the popular QM9 benchmark dataset, and we discuss the broader relevance of these ideas to equivariant geometric machine-learning.
Fast evaluation of real spherical harmonics and their derivatives in Cartesian coordinates
Feb 16, 2023Spherical harmonics provide a smooth, orthogonal, and symmetry-adapted basis to expand functions on a sphere, and they are used routinely in computer graphics, signal processing and different fields of science, from geology to quantum chemistry. More recently, spherical harmonics have become a key component of rotationally equivariant models for geometric deep learning, where they are used in combination with distance-dependent functions to describe the distribution of neighbors within local spherical environments within a point cloud. We present a fast and elegant algorithm for the evaluation of the real-valued spherical harmonics. Our construction integrates many of the desirable features of existing schemes and allows to compute Cartesian derivatives in a numerically stable and computationally efficient manner. We provide an efficient C implementation of the proposed algorithm, along with easy-to-use Python bindings.
A smooth basis for atomistic machine learning
Sep 05, 2022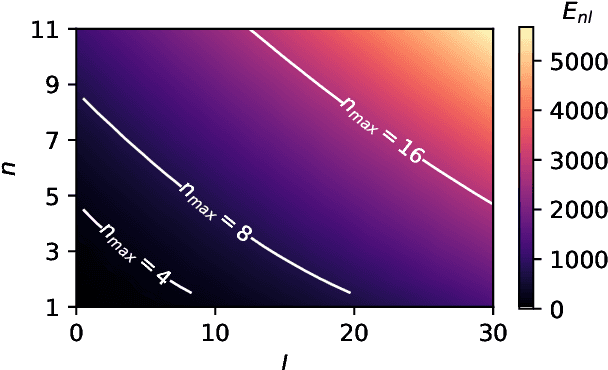
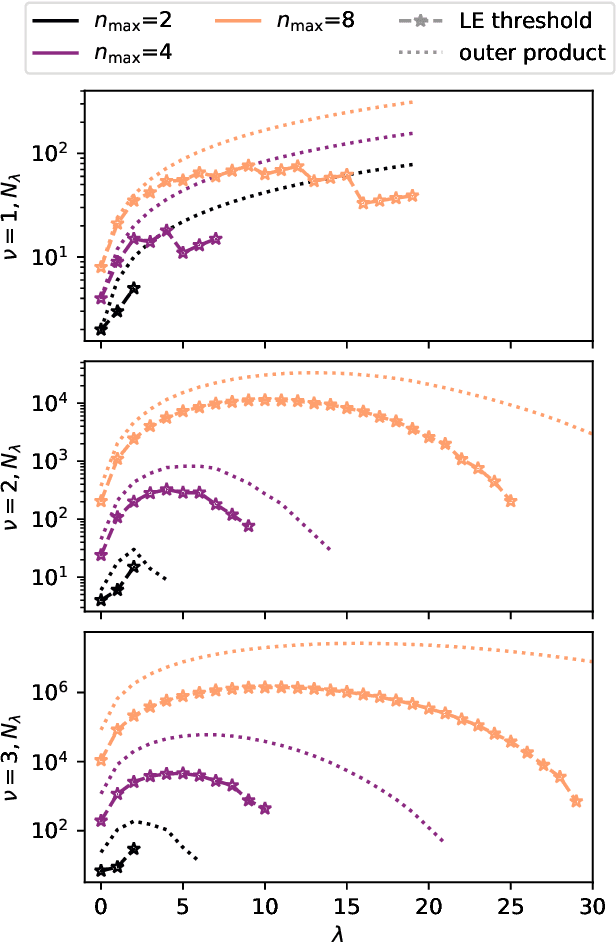
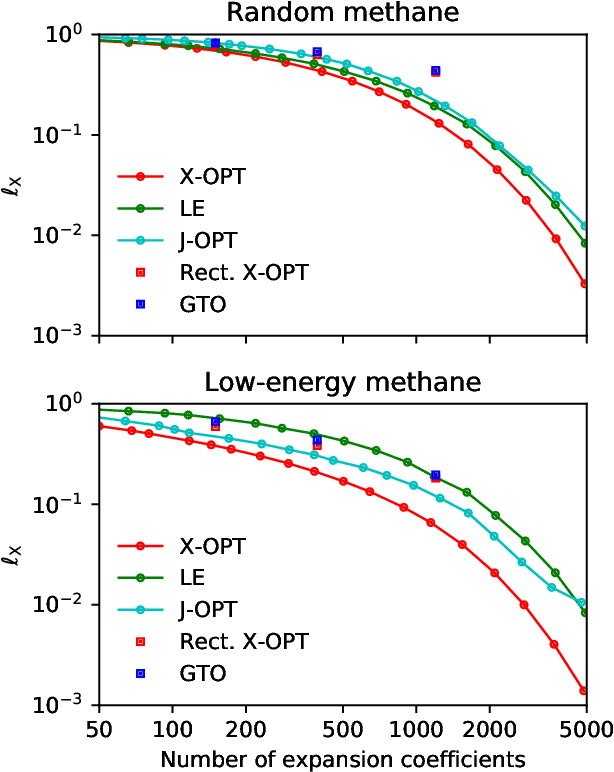

Machine learning frameworks based on correlations of interatomic positions begin with a discretized description of the density of other atoms in the neighbourhood of each atom in the system. Symmetry considerations support the use of spherical harmonics to expand the angular dependence of this density, but there is as yet no clear rationale to choose one radial basis over another. Here we investigate the basis that results from the solution of the Laplacian eigenvalue problem within a sphere around the atom of interest. We show that this generates the smoothest possible basis of a given size within the sphere, and that a tensor product of Laplacian eigenstates also provides the smoothest possible basis for expanding any higher-order correlation of the atomic density within the appropriate hypersphere. We consider several unsupervised metrics of the quality of a basis for a given dataset, and show that the Laplacian eigenstate basis has a performance that is much better than some widely used basis sets and is competitive with data-driven bases that numerically optimize each metric. In supervised machine learning tests, we find that the optimal function smoothness of the Laplacian eigenstates leads to comparable or better performance than can be obtained from a data-driven basis of a similar size that has been optimized to describe the atom-density correlation for the specific dataset. We conclude that the smoothness of the basis functions is a key and hitherto largely overlooked aspect of successful atomic density representations.