 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeA smooth basis for atomistic machine learning
Paper and Code
Sep 05, 2022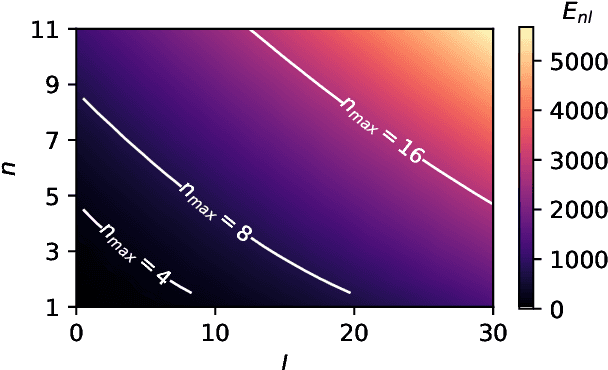
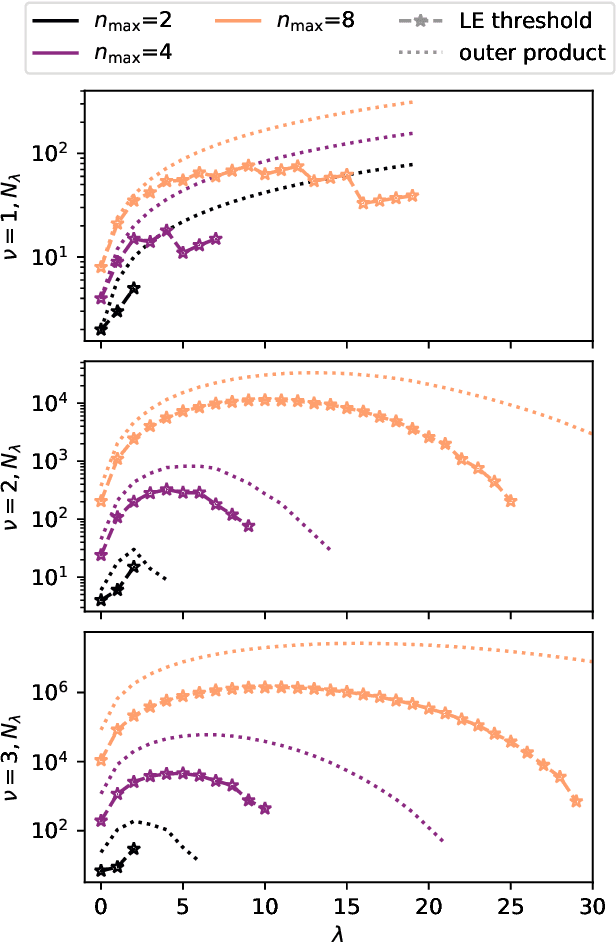
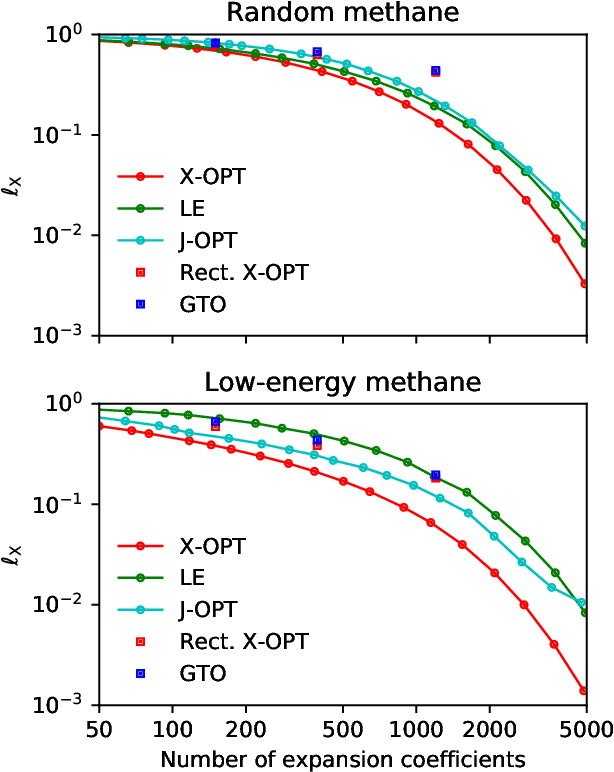

Machine learning frameworks based on correlations of interatomic positions begin with a discretized description of the density of other atoms in the neighbourhood of each atom in the system. Symmetry considerations support the use of spherical harmonics to expand the angular dependence of this density, but there is as yet no clear rationale to choose one radial basis over another. Here we investigate the basis that results from the solution of the Laplacian eigenvalue problem within a sphere around the atom of interest. We show that this generates the smoothest possible basis of a given size within the sphere, and that a tensor product of Laplacian eigenstates also provides the smoothest possible basis for expanding any higher-order correlation of the atomic density within the appropriate hypersphere. We consider several unsupervised metrics of the quality of a basis for a given dataset, and show that the Laplacian eigenstate basis has a performance that is much better than some widely used basis sets and is competitive with data-driven bases that numerically optimize each metric. In supervised machine learning tests, we find that the optimal function smoothness of the Laplacian eigenstates leads to comparable or better performance than can be obtained from a data-driven basis of a similar size that has been optimized to describe the atom-density correlation for the specific dataset. We conclude that the smoothness of the basis functions is a key and hitherto largely overlooked aspect of successful atomic density representations.