 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeUnderstanding the impact of class imbalance on the performance of chest x-ray image classifiers
Dec 23, 2021



This work aims to understand the impact of class imbalance on the performance of chest x-ray classifiers, in light of the standard evaluation practices adopted by researchers in terms of discrimination and calibration performance. Firstly, we conducted a literature study to analyze common scientific practices and confirmed that: (1) even when dealing with highly imbalanced datasets, the community tends to use metrics that are dominated by the majority class; and (2) it is still uncommon to include calibration studies for chest x-ray classifiers, albeit its importance in the context of healthcare. Secondly, we perform a systematic experiment on two major chest x-ray datasets to explore the behavior of several performance metrics under different class ratios and show that widely adopted metrics can conceal the performance in the minority class. Finally, we propose the adoption of two alternative metrics, the precision-recall curve and the Balanced Brier score, which better reflect the performance of the system in such scenarios. Our results indicate that current evaluation practices adopted by the research community for chest x-ray classifiers may not reflect the performance of such systems for computer-aided diagnosis in real clinical scenarios, and suggest alternatives to improve this situation.
Hybrid graph convolutional neural networks for landmark-based anatomical segmentation
Jun 17, 2021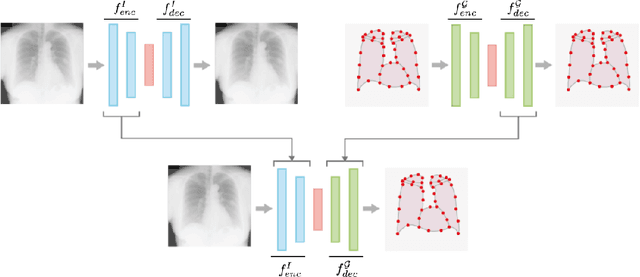
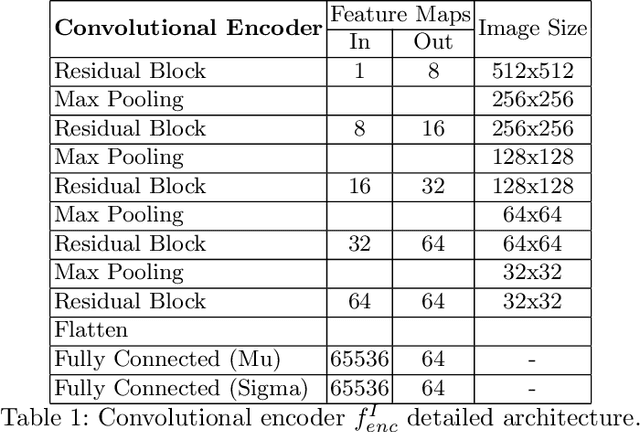
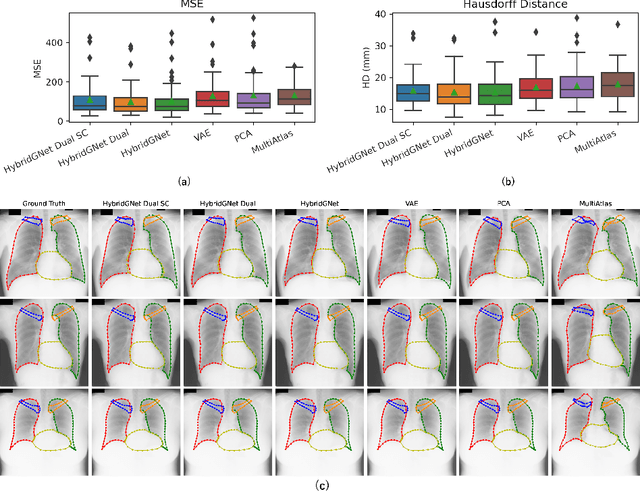
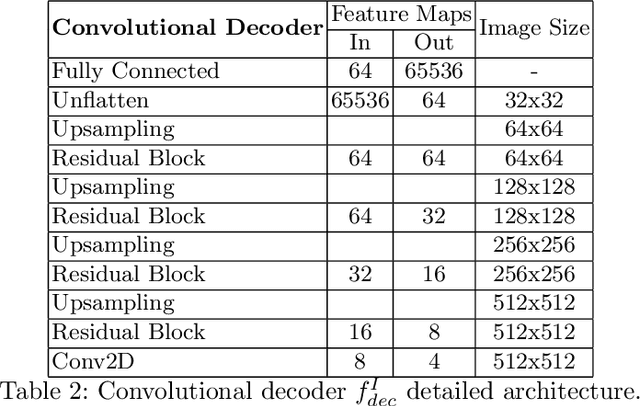
In this work we address the problem of landmark-based segmentation for anatomical structures. We propose HybridGNet, an encoder-decoder neural architecture which combines standard convolutions for image feature encoding, with graph convolutional neural networks to decode plausible representations of anatomical structures. We benchmark the proposed architecture considering other standard landmark and pixel-based models for anatomical segmentation in chest x-ray images, and found that HybridGNet is more robust to image occlusions. We also show that it can be used to construct landmark-based segmentations from pixel level annotations. Our experimental results suggest that HybridGNet produces accurate and anatomically plausible landmark-based segmentations, by naturally incorporating shape constraints within the decoding process via spectral convolutions.
Inferring unknown biological function by integration of GO annotations and gene expression data
Aug 12, 2016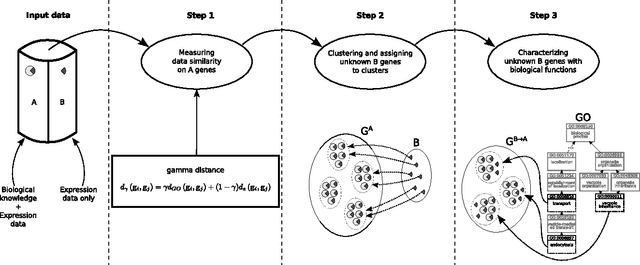
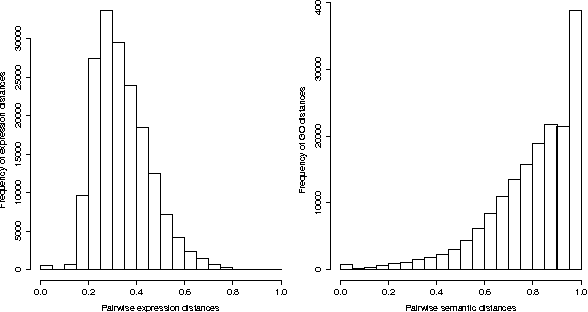

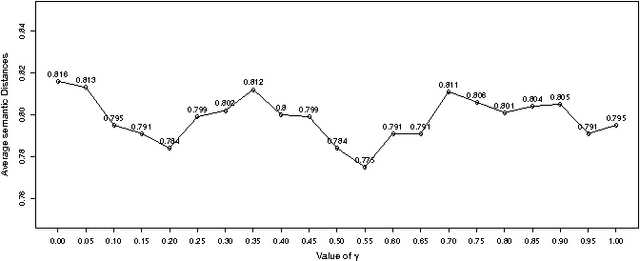
Characterizing genes with semantic information is an important process regarding the description of gene products. In spite that complete genomes of many organisms have been already sequenced, the biological functions of all of their genes are still unknown. Since experimentally studying the functions of those genes, one by one, would be unfeasible, new computational methods for gene functions inference are needed. We present here a novel computational approach for inferring biological function for a set of genes with previously unknown function, given a set of genes with well-known information. This approach is based on the premise that genes with similar behaviour should be grouped together. This is known as the guilt-by-association principle. Thus, it is possible to take advantage of clustering techniques to obtain groups of unknown genes that are co-clustered with genes that have well-known semantic information (GO annotations). Meaningful knowledge to infer unknown semantic information can therefore be provided by these well-known genes. We provide a method to explore the potential function of new genes according to those currently annotated. The results obtained indicate that the proposed approach could be a useful and effective tool when used by biologists to guide the inference of biological functions for recently discovered genes. Our work sets an important landmark in the field of identifying unknown gene functions through clustering, using an external source of biological input. A simple web interface to this proposal can be found at http://fich.unl.edu.ar/sinc/webdemo/gamma-am/.