 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAutomatic Catalog of RRLyrae from $\sim$ 14 million VVV Light Curves: How far can we go with traditional machine-learning?
May 01, 2020



The creation of a 3D map of the bulge using RRLyrae (RRL) is one of the main goals of the VVV(X) surveys. The overwhelming number of sources under analysis request the use of automatic procedures. In this context, previous works introduced the use of Machine Learning (ML) methods for the variable star classification. Our goal is the development and analysis of an automatic procedure, based on ML, for the identification of RRLs in the VVV Survey. This procedure will be use to generate reliable catalogs integrated over several tiles in the survey. After the reconstruction of light-curves, we extract a set of period and intensity-based features. We use for the first time a new subset of pseudo color features. We discuss all the appropriate steps needed to define our automatic pipeline: selection of quality measures; sampling procedures; classifier setup and model selection. As final result, we construct an ensemble classifier with an average Recall of 0.48 and average Precision of 0.86 over 15 tiles. We also make available our processed datasets and a catalog of candidate RRLs. Perhaps most interestingly, from a classification perspective based on photometric broad-band data, is that our results indicate that Color is an informative feature type of the RRL that should be considered for automatic classification methods via ML. We also argue that Recall and Precision in both tables and curves are high quality metrics for this highly imbalanced problem. Furthermore, we show for our VVV data-set that to have good estimates it is important to use the original distribution more than reduced samples with an artificial balance. Finally, we show that the use of ensemble classifiers helps resolve the crucial model selection step, and that most errors in the identification of RRLs are related to low quality observations of some sources or to the difficulty to resolve the RRL-C type given the date.
Inferring unknown biological function by integration of GO annotations and gene expression data
Aug 12, 2016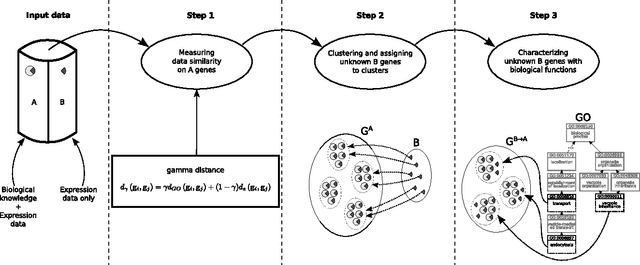
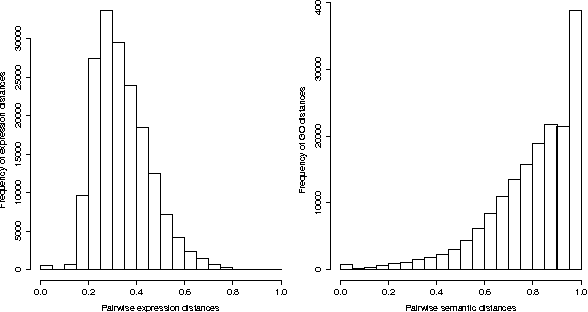

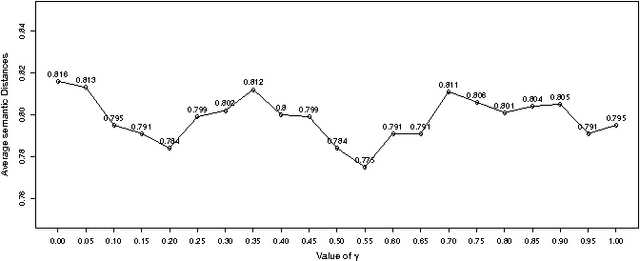
Characterizing genes with semantic information is an important process regarding the description of gene products. In spite that complete genomes of many organisms have been already sequenced, the biological functions of all of their genes are still unknown. Since experimentally studying the functions of those genes, one by one, would be unfeasible, new computational methods for gene functions inference are needed. We present here a novel computational approach for inferring biological function for a set of genes with previously unknown function, given a set of genes with well-known information. This approach is based on the premise that genes with similar behaviour should be grouped together. This is known as the guilt-by-association principle. Thus, it is possible to take advantage of clustering techniques to obtain groups of unknown genes that are co-clustered with genes that have well-known semantic information (GO annotations). Meaningful knowledge to infer unknown semantic information can therefore be provided by these well-known genes. We provide a method to explore the potential function of new genes according to those currently annotated. The results obtained indicate that the proposed approach could be a useful and effective tool when used by biologists to guide the inference of biological functions for recently discovered genes. Our work sets an important landmark in the field of identifying unknown gene functions through clustering, using an external source of biological input. A simple web interface to this proposal can be found at http://fich.unl.edu.ar/sinc/webdemo/gamma-am/.