 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeevolSOM: an R Package for evolutionary conservation analysis with SOMs
Feb 09, 2024Motivation: Unraveling the connection between genes and traits is crucial for solving many biological puzzles. Genes provide instructions for building cellular machinery, directing the processes that sustain life. RNA molecules and proteins, derived from these genetic instructions, play crucial roles in shaping cell structures, influencing reactions, and guiding behavior. This fundamental biological principle links genetic makeup to observable traits, but integrating and extracting meaningful relationships from this complex, multimodal data presents a significant challenge. Results: We introduce evolSOM, a novel R package that utilizes Self-Organizing Maps (SOMs) to explore and visualize the conservation of biological variables, easing the integration of phenotypic and genotypic attributes. By constructing species-specific or condition-specific SOMs that capture non-redundant patterns, evolSOM allows the analysis of displacement of biological variables between species or conditions. Variables displaced together suggest membership in the same regulatory network, and the nature of the displacement may hold biological significance. The package automatically calculates and graphically presents these displacements, enabling efficient comparison and revealing conserved and displaced variables. The package facilitates the integration of diverse phenotypic data types, enabling the exploration of potential gene drivers underlying observed phenotypic changes. Its user-friendly interface and visualization capabilities enhance the accessibility of complex network analyses. Illustratively, we employed evolSOM to study the displacement of genes and phenotypic traits, successfully identifying potential drivers of phenotypic differentiation in grass leaves. Availability: The package is open-source and is available at https://github.com/sanprochetto/evolSOM.
Inferring unknown biological function by integration of GO annotations and gene expression data
Aug 12, 2016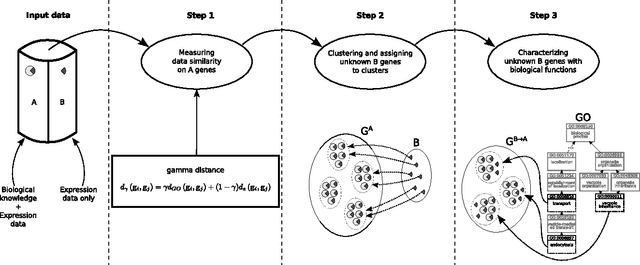
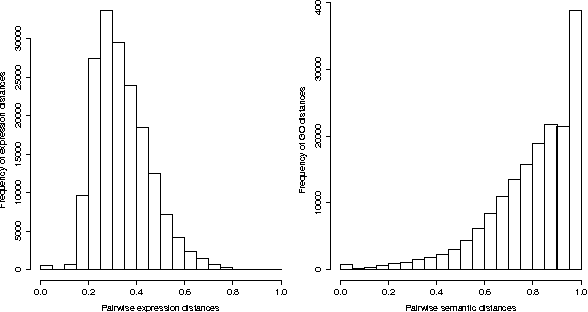

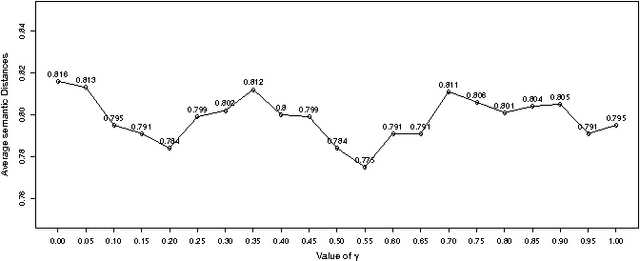
Characterizing genes with semantic information is an important process regarding the description of gene products. In spite that complete genomes of many organisms have been already sequenced, the biological functions of all of their genes are still unknown. Since experimentally studying the functions of those genes, one by one, would be unfeasible, new computational methods for gene functions inference are needed. We present here a novel computational approach for inferring biological function for a set of genes with previously unknown function, given a set of genes with well-known information. This approach is based on the premise that genes with similar behaviour should be grouped together. This is known as the guilt-by-association principle. Thus, it is possible to take advantage of clustering techniques to obtain groups of unknown genes that are co-clustered with genes that have well-known semantic information (GO annotations). Meaningful knowledge to infer unknown semantic information can therefore be provided by these well-known genes. We provide a method to explore the potential function of new genes according to those currently annotated. The results obtained indicate that the proposed approach could be a useful and effective tool when used by biologists to guide the inference of biological functions for recently discovered genes. Our work sets an important landmark in the field of identifying unknown gene functions through clustering, using an external source of biological input. A simple web interface to this proposal can be found at http://fich.unl.edu.ar/sinc/webdemo/gamma-am/.