 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to Edgesc-OTGM: Single-Cell Perturbation Modeling by Solving Optimal Mass Transport on the Manifold of Gaussian Mixtures
May 06, 2024
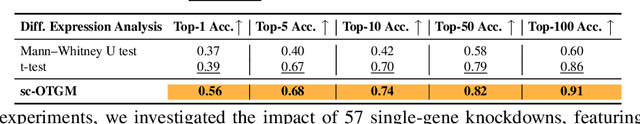


Influenced by breakthroughs in LLMs, single-cell foundation models are emerging. While these models show successful performance in cell type clustering, phenotype classification, and gene perturbation response prediction, it remains to be seen if a simpler model could achieve comparable or better results, especially with limited data. This is important, as the quantity and quality of single-cell data typically fall short of the standards in textual data used for training LLMs. Single-cell sequencing often suffers from technical artifacts, dropout events, and batch effects. These challenges are compounded in a weakly supervised setting, where the labels of cell states can be noisy, further complicating the analysis. To tackle these challenges, we present sc-OTGM, streamlined with less than 500K parameters, making it approximately 100x more compact than the foundation models, offering an efficient alternative. sc-OTGM is an unsupervised model grounded in the inductive bias that the scRNAseq data can be generated from a combination of the finite multivariate Gaussian distributions. The core function of sc-OTGM is to create a probabilistic latent space utilizing a GMM as its prior distribution and distinguish between distinct cell populations by learning their respective marginal PDFs. It uses a Hit-and-Run Markov chain sampler to determine the OT plan across these PDFs within the GMM framework. We evaluated our model against a CRISPR-mediated perturbation dataset, called CROP-seq, consisting of 57 one-gene perturbations. Our results demonstrate that sc-OTGM is effective in cell state classification, aids in the analysis of differential gene expression, and ranks genes for target identification through a recommender system. It also predicts the effects of single-gene perturbations on downstream gene regulation and generates synthetic scRNA-seq data conditioned on specific cell states.
SE(3)-Invariant Multiparameter Persistent Homology for Chiral-Sensitive Molecular Property Prediction
Dec 12, 2023



In this study, we present a novel computational method for generating molecular fingerprints using multiparameter persistent homology (MPPH). This technique holds considerable significance for drug discovery and materials science, where precise molecular property prediction is vital. By integrating SE(3)-invariance with Vietoris-Rips persistent homology, we effectively capture the three-dimensional representations of molecular chirality. This non-superimposable mirror image property directly influences the molecular interactions, serving as an essential factor in molecular property prediction. We explore the underlying topologies and patterns in molecular structures by applying Vietoris-Rips persistent homology across varying scales and parameters such as atomic weight, partial charge, bond type, and chirality. Our method's efficacy can be improved by incorporating additional parameters such as aromaticity, orbital hybridization, bond polarity, conjugated systems, as well as bond and torsion angles. Additionally, we leverage Stochastic Gradient Langevin Boosting in a Bayesian ensemble of GBDTs to obtain aleatoric and epistemic uncertainty estimates for gradient boosting models. With these uncertainty estimates, we prioritize high-uncertainty samples for active learning and model fine-tuning, benefiting scenarios where data labeling is costly or time consuming. Compared to conventional GNNs which usually suffer from oversmoothing and oversquashing, MPPH provides a more comprehensive and interpretable characterization of molecular data topology. We substantiate our approach with theoretical stability guarantees and demonstrate its superior performance over existing state-of-the-art methods in predicting molecular properties through extensive evaluations on the MoleculeNet benchmark datasets.
Multiparameter Persistent Homology for Molecular Property Prediction
Nov 17, 2023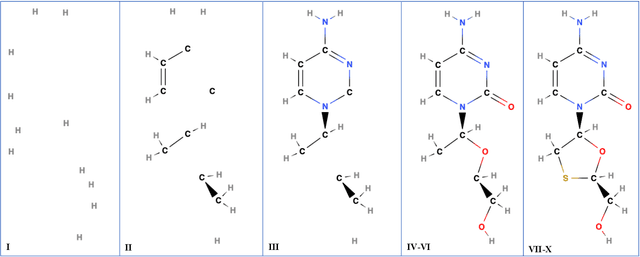

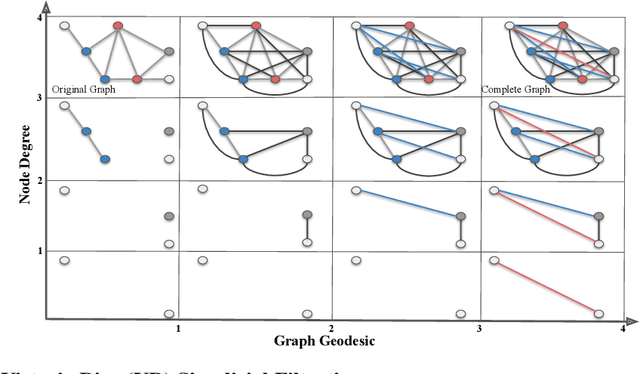

In this study, we present a novel molecular fingerprint generation method based on multiparameter persistent homology. This approach reveals the latent structures and relationships within molecular geometry, and detects topological features that exhibit persistence across multiple scales along multiple parameters, such as atomic mass, partial charge, and bond type, and can be further enhanced by incorporating additional parameters like ionization energy, electron affinity, chirality and orbital hybridization. The proposed fingerprinting method provides fresh perspectives on molecular structure that are not easily discernible from single-parameter or single-scale analysis. Besides, in comparison with traditional graph neural networks, multiparameter persistent homology has the advantage of providing a more comprehensive and interpretable characterization of the topology of the molecular data. We have established theoretical stability guarantees for multiparameter persistent homology, and have conducted extensive experiments on the Lipophilicity, FreeSolv, and ESOL datasets to demonstrate its effectiveness in predicting molecular properties.
Topology-Aware Focal Loss for 3D Image Segmentation
Apr 27, 2023The efficacy of segmentation algorithms is frequently compromised by topological errors like overlapping regions, disrupted connections, and voids. To tackle this problem, we introduce a novel loss function, namely Topology-Aware Focal Loss (TAFL), that incorporates the conventional Focal Loss with a topological constraint term based on the Wasserstein distance between the ground truth and predicted segmentation masks' persistence diagrams. By enforcing identical topology as the ground truth, the topological constraint can effectively resolve topological errors, while Focal Loss tackles class imbalance. We begin by constructing persistence diagrams from filtered cubical complexes of the ground truth and predicted segmentation masks. We subsequently utilize the Sinkhorn-Knopp algorithm to determine the optimal transport plan between the two persistence diagrams. The resultant transport plan minimizes the cost of transporting mass from one distribution to the other and provides a mapping between the points in the two persistence diagrams. We then compute the Wasserstein distance based on this travel plan to measure the topological dissimilarity between the ground truth and predicted masks. We evaluate our approach by training a 3D U-Net with the MICCAI Brain Tumor Segmentation (BraTS) challenge validation dataset, which requires accurate segmentation of 3D MRI scans that integrate various modalities for the precise identification and tracking of malignant brain tumors. Then, we demonstrate that the quality of segmentation performance is enhanced by regularizing the focal loss through the addition of a topological constraint as a penalty term.
EEG-NeXt: A Modernized ConvNet for The Classification of Cognitive Activity from EEG
Dec 08, 2022One of the main challenges in electroencephalogram (EEG) based brain-computer interface (BCI) systems is learning the subject/session invariant features to classify cognitive activities within an end-to-end discriminative setting. We propose a novel end-to-end machine learning pipeline, EEG-NeXt, which facilitates transfer learning by: i) aligning the EEG trials from different subjects in the Euclidean-space, ii) tailoring the techniques of deep learning for the scalograms of EEG signals to capture better frequency localization for low-frequency, longer-duration events, and iii) utilizing pretrained ConvNeXt (a modernized ResNet architecture which supersedes state-of-the-art (SOTA) image classification models) as the backbone network via adaptive finetuning. On publicly available datasets (Physionet Sleep Cassette and BNCI2014001) we benchmark our method against SOTA via cross-subject validation and demonstrate improved accuracy in cognitive activity classification along with better generalizability across cohorts.
ToDD: Topological Compound Fingerprinting in Computer-Aided Drug Discovery
Nov 07, 2022In computer-aided drug discovery (CADD), virtual screening (VS) is used for identifying the drug candidates that are most likely to bind to a molecular target in a large library of compounds. Most VS methods to date have focused on using canonical compound representations (e.g., SMILES strings, Morgan fingerprints) or generating alternative fingerprints of the compounds by training progressively more complex variational autoencoders (VAEs) and graph neural networks (GNNs). Although VAEs and GNNs led to significant improvements in VS performance, these methods suffer from reduced performance when scaling to large virtual compound datasets. The performance of these methods has shown only incremental improvements in the past few years. To address this problem, we developed a novel method using multiparameter persistence (MP) homology that produces topological fingerprints of the compounds as multidimensional vectors. Our primary contribution is framing the VS process as a new topology-based graph ranking problem by partitioning a compound into chemical substructures informed by the periodic properties of its atoms and extracting their persistent homology features at multiple resolution levels. We show that the margin loss fine-tuning of pretrained Triplet networks attains highly competitive results in differentiating between compounds in the embedding space and ranking their likelihood of becoming effective drug candidates. We further establish theoretical guarantees for the stability properties of our proposed MP signatures, and demonstrate that our models, enhanced by the MP signatures, outperform state-of-the-art methods on benchmark datasets by a wide and highly statistically significant margin (e.g., 93% gain for Cleves-Jain and 54% gain for DUD-E Diverse dataset).