 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to Edgesc-OTGM: Single-Cell Perturbation Modeling by Solving Optimal Mass Transport on the Manifold of Gaussian Mixtures
Paper and Code

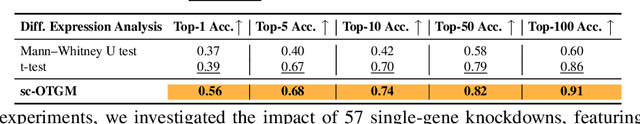


Influenced by breakthroughs in LLMs, single-cell foundation models are emerging. While these models show successful performance in cell type clustering, phenotype classification, and gene perturbation response prediction, it remains to be seen if a simpler model could achieve comparable or better results, especially with limited data. This is important, as the quantity and quality of single-cell data typically fall short of the standards in textual data used for training LLMs. Single-cell sequencing often suffers from technical artifacts, dropout events, and batch effects. These challenges are compounded in a weakly supervised setting, where the labels of cell states can be noisy, further complicating the analysis. To tackle these challenges, we present sc-OTGM, streamlined with less than 500K parameters, making it approximately 100x more compact than the foundation models, offering an efficient alternative. sc-OTGM is an unsupervised model grounded in the inductive bias that the scRNAseq data can be generated from a combination of the finite multivariate Gaussian distributions. The core function of sc-OTGM is to create a probabilistic latent space utilizing a GMM as its prior distribution and distinguish between distinct cell populations by learning their respective marginal PDFs. It uses a Hit-and-Run Markov chain sampler to determine the OT plan across these PDFs within the GMM framework. We evaluated our model against a CRISPR-mediated perturbation dataset, called CROP-seq, consisting of 57 one-gene perturbations. Our results demonstrate that sc-OTGM is effective in cell state classification, aids in the analysis of differential gene expression, and ranks genes for target identification through a recommender system. It also predicts the effects of single-gene perturbations on downstream gene regulation and generates synthetic scRNA-seq data conditioned on specific cell states.