 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeGraphRAG for Engineering Diagrams: ChatP&ID Enables LLM Interaction with P&IDs
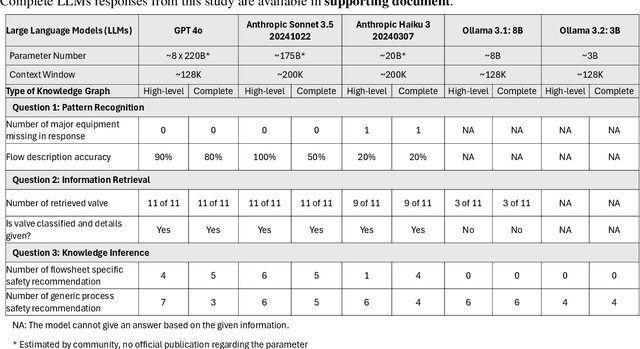
Mar 23, 2026Large Language Models (LLMs) combined with Retrieval-Augmented Generation (RAG) and knowledge graphs offer new opportunities for interacting with engineering diagrams such as Piping and Instrumentation Diagrams (P&IDs). However, directly processing raw images or smart P&ID files with LLMs is often costly, inefficient, and prone to hallucinations. This work introduces ChatP&ID, an agentic framework that enables grounded and cost-effective natural-language interaction with P&IDs using Graph Retrieval-Augmented Generation (GraphRAG), a paradigm we refer to as GraphRAG for engineering diagrams. Smart P&IDs encoded in the DEXPI standard are transformed into structured knowledge graphs, which serve as the basis for graph-based retrieval and reasoning by LLM agents. This approach enables reliable querying of engineering diagrams while significantly reducing computational cost. Benchmarking across commercial LLM APIs (OpenAI, Anthropic) demonstrates that graph-based representations improve accuracy by 18% over raw image inputs and reduce token costs by 85% compared to directly ingesting smart P&ID files. While small open-source models still struggle to interpret knowledge graph formats and structured engineering data, integrating them with VectorRAG and PathRAG improves response accuracy by up to 40%. Notably, GPT-5-mini combined with ContextRAG achieves 91% accuracy at a cost of only $0.004 per task. The resulting ChatP&ID interface enables intuitive natural-language interaction with complex engineering diagrams and lays the groundwork for AI-assisted process engineering tasks such as Hazard and Operability Studies (HAZOP) and multi-agent analysis.
Grounding Large Language Models in Reaction Knowledge Graphs for Synthesis Retrieval
Jan 22, 2026Large Language Models (LLMs) can aid synthesis planning in chemistry, but standard prompting methods often yield hallucinated or outdated suggestions. We study LLM interactions with a reaction knowledge graph by casting reaction path retrieval as a Text2Cypher (natural language to graph query) generation problem, and define single- and multi-step retrieval tasks. We compare zero-shot prompting to one-shot variants using static, random, and embedding-based exemplar selection, and assess a checklist-driven validator/corrector loop. To evaluate our framework, we consider query validity and retrieval accuracy. We find that one-shot prompting with aligned exemplars consistently performs best. Our checklist-style self-correction loop mainly improves executability in zero-shot settings and offers limited additional retrieval gains once a good exemplar is present. We provide a reproducible Text2Cypher evaluation setup to facilitate further work on KG-grounded LLMs for synthesis planning. Code is available at https://github.com/Intelligent-molecular-systems/KG-LLM-Synthesis-Retrieval.
Talking like Piping and Instrumentation Diagrams (P&IDs)
Feb 26, 2025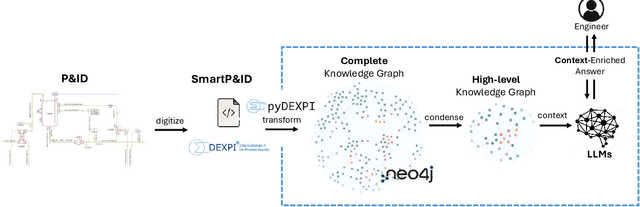
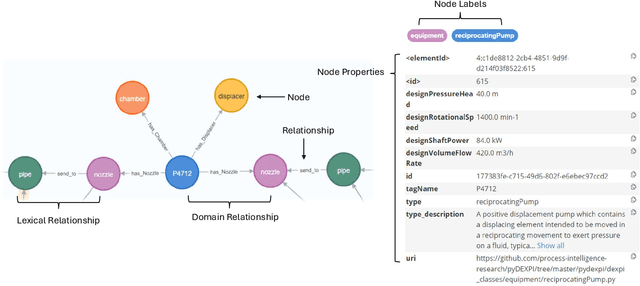
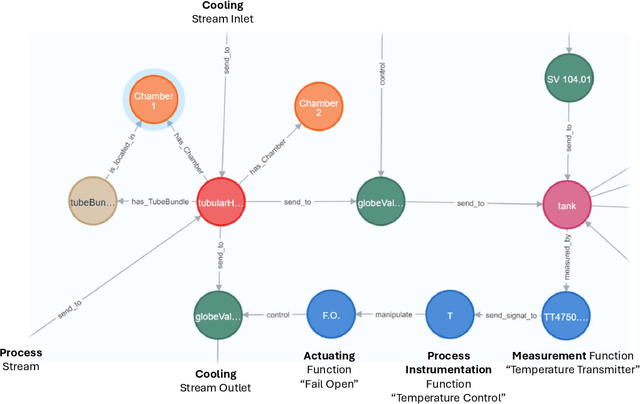
We propose a methodology that allows communication with Piping and Instrumentation Diagrams (P&IDs) using natural language. In particular, we represent P&IDs through the DEXPI data model as labeled property graphs and integrate them with Large Language Models (LLMs). The approach consists of three main parts: 1) P&IDs are cast into a graph representation from the DEXPI format using our pyDEXPI Python package. 2) A tool for generating P&ID knowledge graphs from pyDEXPI. 3) Integration of the P&ID knowledge graph to LLMs using graph-based retrieval augmented generation (graph-RAG). This approach allows users to communicate with P&IDs using natural language. It extends LLM's ability to retrieve contextual data from P&IDs and mitigate hallucinations. Leveraging the LLM's large corpus, the model is also able to interpret process information in PIDs, which could help engineers in their daily tasks. In the future, this work will also open up opportunities in the context of other generative Artificial Intelligence (genAI) solutions on P&IDs, and AI-assisted HAZOP studies.
Rule-based autocorrection of Piping and Instrumentation Diagrams (P&IDs) on graphs
Feb 18, 2025A piping and instrumentation diagram (P&ID) is a central reference document in chemical process engineering. Currently, chemical engineers manually review P&IDs through visual inspection to find and rectify errors. However, engineering projects can involve hundreds to thousands of P&ID pages, creating a significant revision workload. This study proposes a rule-based method to support engineers with error detection and correction in P&IDs. The method is based on a graph representation of P&IDs, enabling automated error detection and correction, i.e., autocorrection, through rule graphs. We use our pyDEXPI Python package to generate P&ID graphs from DEXPI-standard P&IDs. In this study, we developed 33 rules based on chemical engineering knowledge and heuristics, with five selected rules demonstrated as examples. A case study on an illustrative P&ID validates the reliability and effectiveness of the rule-based autocorrection method in revising P&IDs.
ENFORCE: Exact Nonlinear Constrained Learning with Adaptive-depth Neural Projection
Feb 10, 2025



Ensuring neural networks adhere to domain-specific constraints is crucial for addressing safety and ethical concerns while also enhancing prediction accuracy. Despite the nonlinear nature of most real-world tasks, existing methods are predominantly limited to affine or convex constraints. We introduce ENFORCE, a neural network architecture that guarantees predictions to satisfy nonlinear constraints exactly. ENFORCE is trained with standard unconstrained gradient-based optimizers (e.g., Adam) and leverages autodifferentiation and local neural projections to enforce any $\mathcal{C}^1$ constraint to arbitrary tolerance $\epsilon$. We build an adaptive-depth neural projection (AdaNP) module that dynamically adjusts its complexity to suit the specific problem and the required tolerance levels. ENFORCE guarantees satisfaction of equality constraints that are nonlinear in both inputs and outputs of the neural network with minimal (and adjustable) computational cost.
Picard-KKT-hPINN: Enforcing Nonlinear Enthalpy Balances for Physically Consistent Neural Networks
Jan 29, 2025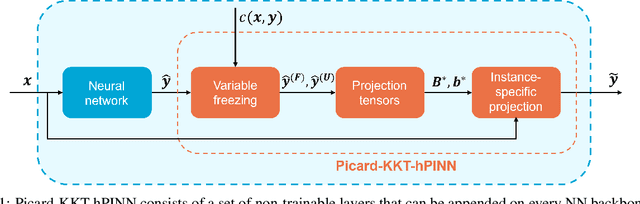
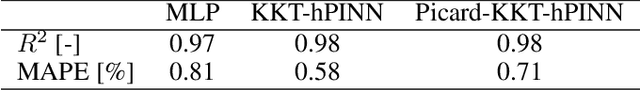
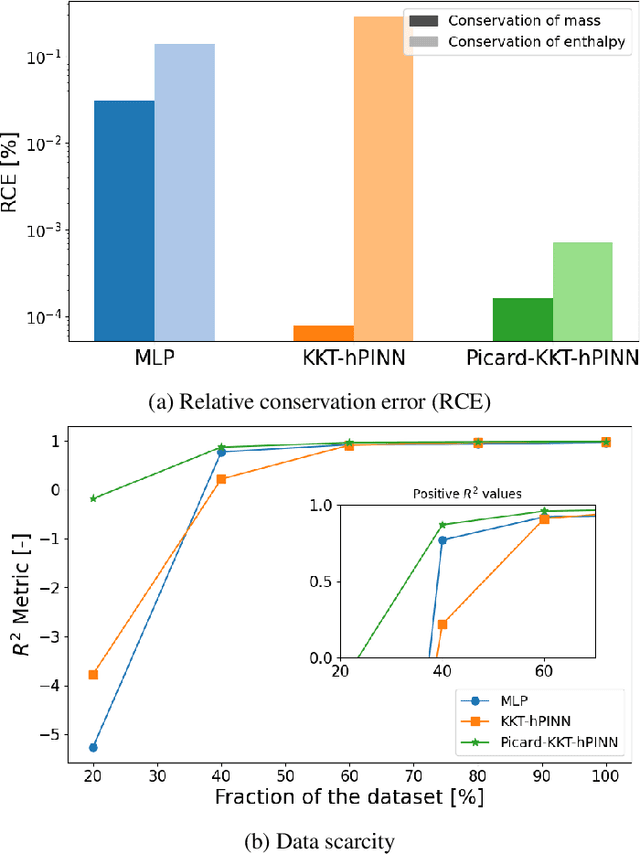
Neural networks are widely used as surrogate models but they do not guarantee physically consistent predictions thereby preventing adoption in various applications. We propose a method that can enforce NNs to satisfy physical laws that are nonlinear in nature such as enthalpy balances. Our approach, inspired by Picard successive approximations method, aims to enforce multiplicatively separable constraints by sequentially freezing and projecting a set of the participating variables. We demonstrate our PicardKKThPINN for surrogate modeling of a catalytic packed bed reactor for methanol synthesis. Our results show that the method efficiently enforces nonlinear enthalpy and linear atomic balances at machine-level precision. Additionally, we show that enforcing conservation laws can improve accuracy in data-scarce conditions compared to vanilla multilayer perceptron.
Graph-to-SFILES: Control structure prediction from process topologies using generative artificial intelligence
Nov 30, 2024



Control structure design is an important but tedious step in P&ID development. Generative artificial intelligence (AI) promises to reduce P&ID development time by supporting engineers. Previous research on generative AI in chemical process design mainly represented processes by sequences. However, graphs offer a promising alternative because of their permutation invariance. We propose the Graph-to-SFILES model, a generative AI method to predict control structures from flowsheet topologies. The Graph-to-SFILES model takes the flowsheet topology as a graph input and returns a control-extended flowsheet as a sequence in the SFILES 2.0 notation. We compare four different graph encoder architectures, one of them being a graph neural network (GNN) proposed in this work. The Graph-to-SFILES model achieves a top-5 accuracy of 73.2% when trained on 10,000 flowsheet topologies. In addition, the proposed GNN performs best among the encoder architectures. Compared to a purely sequence-based approach, the Graph-to-SFILES model improves the top-5 accuracy for a relatively small training dataset of 1,000 flowsheets from 0.9% to 28.4%. However, the sequence-based approach performs better on a large-scale dataset of 100,000 flowsheets. These results highlight the potential of graph-based AI models to accelerate P&ID development in small-data regimes but their effectiveness on industry relevant case studies still needs to be investigated.
Bayesian Uncertainty Estimation by Hamiltonian Monte Carlo: Applications to Cardiac MRI Segmentation
Mar 04, 2024Deep learning (DL)-based methods have achieved state-of-the-art performance for a wide range of medical image segmentation tasks. Nevertheless, recent studies show that deep neural networks (DNNs) can be miscalibrated and overconfident, leading to "silent failures" that are risky} for clinical applications. Bayesian statistics provide an intuitive approach to DL failure detection, based on posterior probability estimation. However, Bayesian DL, and in particular the posterior estimation, is intractable for large medical image segmentation DNNs. To tackle this challenge, we propose a Bayesian learning framework by Hamiltonian Monte Carlo (HMC), tempered by cold posterior (CP) to accommodate medical data augmentation, named HMC-CP. For HMC computation, we further propose a cyclical annealing strategy, which captures both local and global geometries of the posterior distribution, enabling highly efficient Bayesian DNN training with the same computational budget requirements as training a single DNN. The resulting Bayesian DNN outputs an ensemble segmentation along with the segmentation uncertainty. We evaluate the proposed HMC-CP extensively on cardiac magnetic resonance image (MRI) segmentation, using in-domain steady-state free precession (SSFP) cine images as well as out-of-domain datasets of quantitative $T_1$ and $T_2$ mapping.
MachineLearnAthon: An Action-Oriented Machine Learning Didactic Concept
Jan 29, 2024Machine Learning (ML) techniques are encountered nowadays across disciplines, from social sciences, through natural sciences to engineering. The broad application of ML and the accelerated pace of its evolution lead to an increasing need for dedicated teaching concepts aimed at making the application of this technology more reliable and responsible. However, teaching ML is a daunting task. Aside from the methodological complexity of ML algorithms, both with respect to theory and implementation, the interdisciplinary and empirical nature of the field need to be taken into consideration. This paper introduces the MachineLearnAthon format, an innovative didactic concept designed to be inclusive for students of different disciplines with heterogeneous levels of mathematics, programming and domain expertise. At the heart of the concept lie ML challenges, which make use of industrial data sets to solve real-world problems. These cover the entire ML pipeline, promoting data literacy and practical skills, from data preparation, through deployment, to evaluation.
Toward autocorrection of chemical process flowsheets using large language models
Dec 05, 2023The process engineering domain widely uses Process Flow Diagrams (PFDs) and Process and Instrumentation Diagrams (P&IDs) to represent process flows and equipment configurations. However, the P&IDs and PFDs, hereafter called flowsheets, can contain errors causing safety hazards, inefficient operation, and unnecessary expenses. Correcting and verifying flowsheets is a tedious, manual process. We propose a novel generative AI methodology for automatically identifying errors in flowsheets and suggesting corrections to the user, i.e., autocorrecting flowsheets. Inspired by the breakthrough of Large Language Models (LLMs) for grammatical autocorrection of human language, we investigate LLMs for the autocorrection of flowsheets. The input to the model is a potentially erroneous flowsheet and the output of the model are suggestions for a corrected flowsheet. We train our autocorrection model on a synthetic dataset in a supervised manner. The model achieves a top-1 accuracy of 80% and a top-5 accuracy of 84% on an independent test dataset of synthetically generated flowsheets. The results suggest that the model can learn to autocorrect the synthetic flowsheets. We envision that flowsheet autocorrection will become a useful tool for chemical engineers.