 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeREP: Predicting the Time-Course of Drug Sensitivity
Jul 27, 2019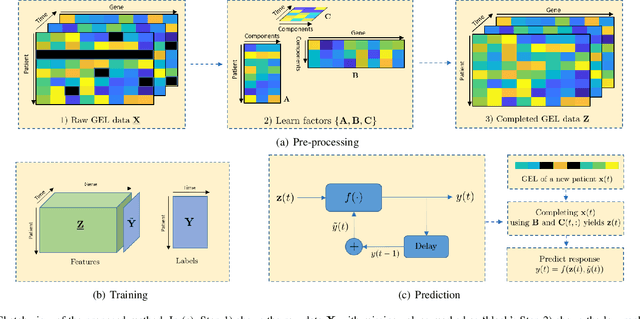
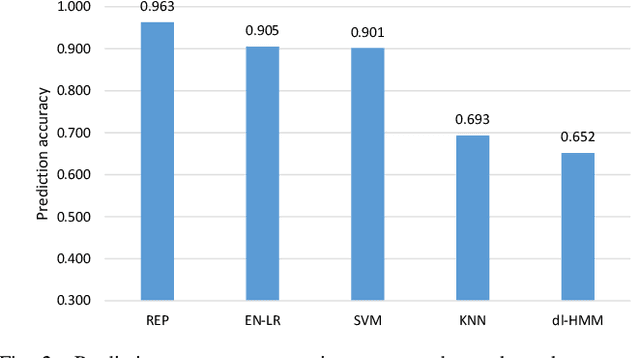

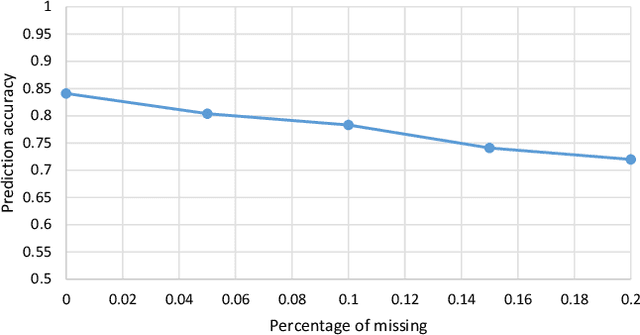
The biological processes involved in a drug's mechanisms of action are oftentimes dynamic, complex and difficult to discern. Time-course gene expression data is a rich source of information that can be used to unravel these complex processes, identify biomarkers of drug sensitivity and predict the response to a drug. However, the majority of previous work has not fully utilized this temporal dimension. In these studies, the gene expression data is either considered at one time-point (before the administration of the drug) or two timepoints (before and after the administration of the drug). This is clearly inadequate in modeling dynamic gene-drug interactions, especially for applications such as long-term drug therapy. In this work, we present a novel REcursive Prediction (REP) framework for drug response prediction by taking advantage of time-course gene expression data. Our goal is to predict drug response values at every stage of a long-term treatment, given the expression levels of genes collected in the previous time-points. To this end, REP employs a built-in recursive structure that exploits the intrinsic time-course nature of the data and integrates past values of drug responses for subsequent predictions. It also incorporates tensor completion that can not only alleviate the impact of noise and missing data, but also predict unseen gene expression levels (GELs). These advantages enable REP to estimate drug response at any stage of a given treatment from some GELs measured in the beginning of the treatment. Extensive experiments on a dataset corresponding to 53 multiple sclerosis patients treated with interferon are included to showcase the effectiveness of REP.
From Gene Expression to Drug Response: A Collaborative Filtering Approach
Oct 31, 2018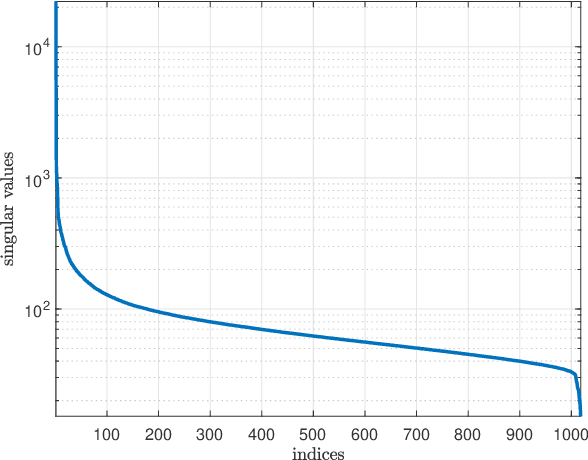
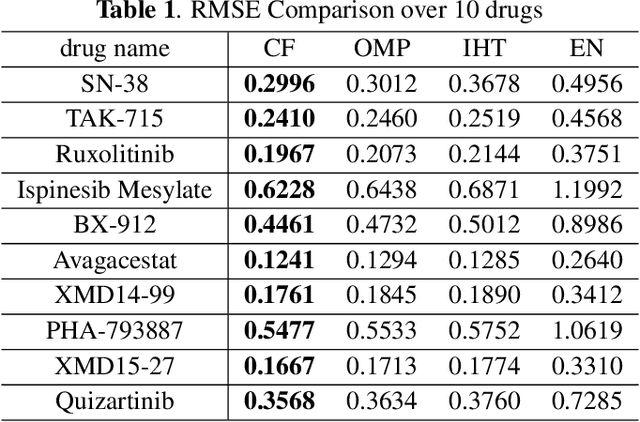
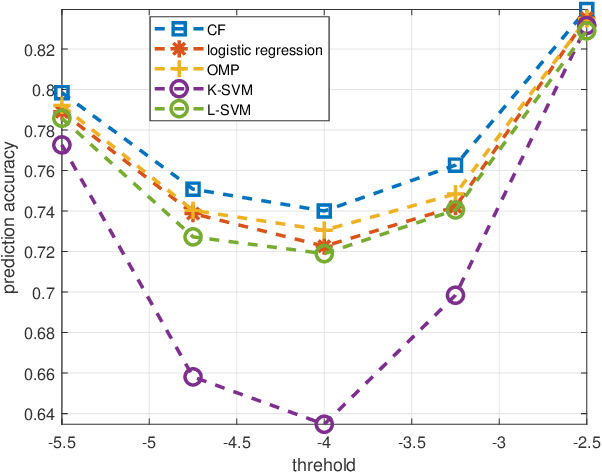
Predicting the response of cancer cells to drugs is an important problem in pharmacogenomics. Recent efforts in generation of large scale datasets profiling gene expression and drug sensitivity in cell lines have provided a unique opportunity to study this problem. However, one major challenge is the small number of samples (cell lines) compared to the number of features (genes) even in these large datasets. We propose a collaborative filtering (CF) like algorithm for modeling gene-drug relationship to identify patients most likely to benefit from a treatment. Due to the correlation of gene expressions in different cell lines, the gene expression matrix is approximately low-rank, which suggests that drug responses could be estimated from a reduced dimension latent space of the gene expression. Towards this end, we propose a joint low-rank matrix factorization and latent linear regression approach. Experiments with data from the Genomics of Drug Sensitivity in Cancer database are included to show that the proposed method can predict drug-gene associations better than the state-of-the-art methods.
A new correlation clustering method for cancer mutation analysis
Jan 25, 2016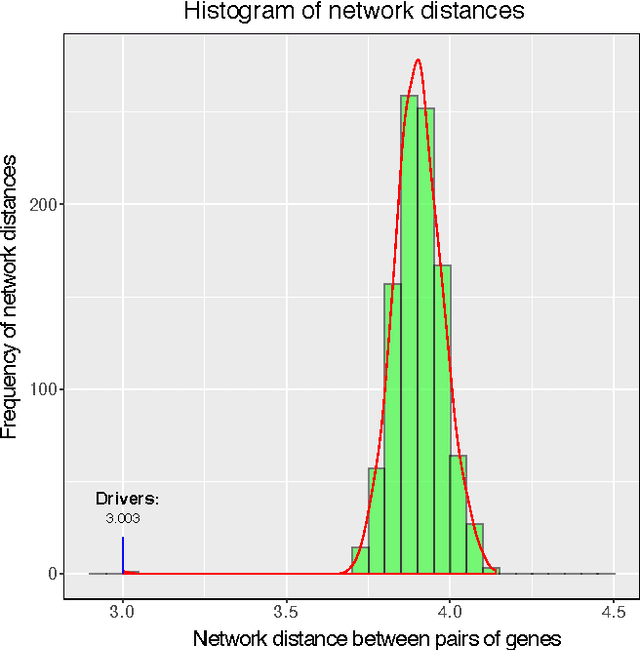
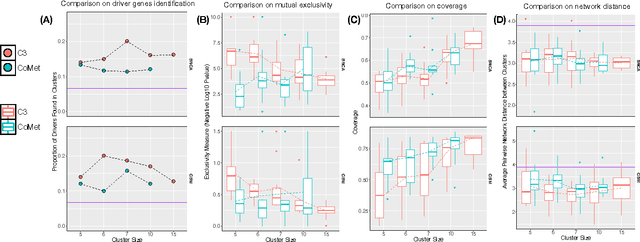
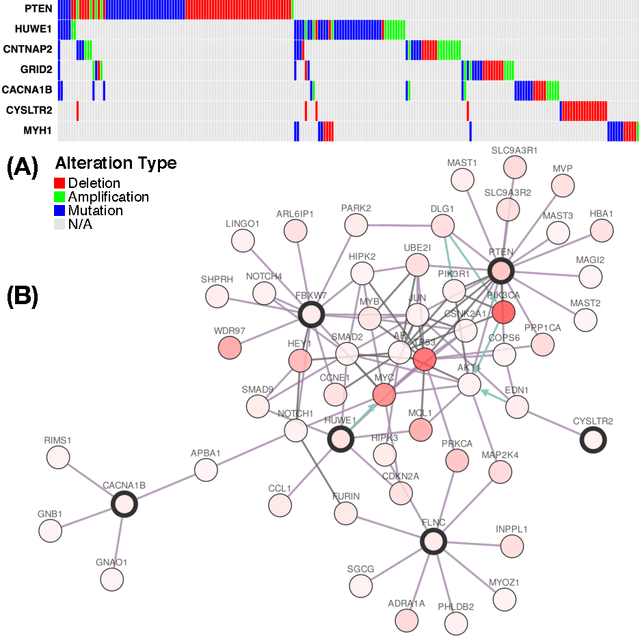
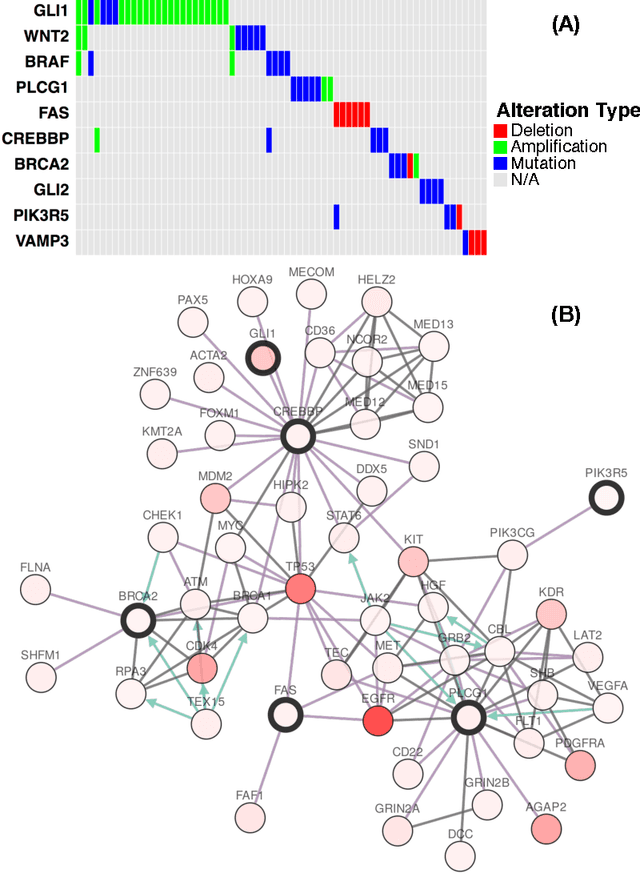
Cancer genomes exhibit a large number of different alterations that affect many genes in a diverse manner. It is widely believed that these alterations follow combinatorial patterns that have a strong connection with the underlying molecular interaction networks and functional pathways. A better understanding of the generative mechanisms behind the mutation rules and their influence on gene communities is of great importance for the process of driver mutations discovery and for identification of network modules related to cancer development and progression. We developed a new method for cancer mutation pattern analysis based on a constrained form of correlation clustering. Correlation clustering is an agnostic learning method that can be used for general community detection problems in which the number of communities or their structure is not known beforehand. The resulting algorithm, named $C^3$, leverages mutual exclusivity of mutations, patient coverage, and driver network concentration principles; it accepts as its input a user determined combination of heterogeneous patient data, such as that available from TCGA (including mutation, copy number, and gene expression information), and creates a large number of clusters containing mutually exclusive mutated genes in a particular type of cancer. The cluster sizes may be required to obey some useful soft size constraints, without impacting the computational complexity of the algorithm. To test $C^3$, we performed a detailed analysis on TCGA breast cancer and glioblastoma data and showed that our algorithm outperforms the state-of-the-art CoMEt method in terms of discovering mutually exclusive gene modules and identifying driver genes. Our $C^3$ method represents a unique tool for efficient and reliable identification of mutation patterns and driver pathways in large-scale cancer genomics studies.